| * Correspondence to: Ryan W. Schmidt, Department of Anthropology, University of Montana, Missoula, Social Science Bldg., 32 Campus Drive, Missoula, MT 59812, USA. E-mail: ryan2.schmidt@umontana.edu Published online 24 July 2010 in J-STAGE (www.jstage.jst.go.jp) DOI: 10.1537/ase.100128 |
In this study, we investigate craniometric variability of Chinese immigrants living in the United States during the late 19th and early 20th centuries. The goal is to determine the geographic origin of these immigrants by examining patterns of biological variation using a quantitative genetic approach. Chinese immigrants who inhabited parts of North America during the building and expansion of the United States in the late 1800s and early 1900s have not been as thoroughly investigated as other groups living in this period, especially in an archaeological context. Here, we have the opportunity to elucidate some important questions using data to which historians usually do not have direct access. That is, the skeletal remains themselves can answer important questions surrounding Chinese immigrant origins and migration to North America.
It is well documented (Chung and Wegars, 2005) that many Chinese immigrants to North America had their remains returned to China for reburial. However, there are historical records of small cemeteries and internments from various towns across the United States. It is these skeletal remains that have the potential to provide insight into migration origins of at least some of the earliest Chinese immigrants to this country. This study involves the analysis of skeletal remains from Carlin, Nevada, a small cemetery excavated in 1996, and the analysis of a craniometric sample from Kodiak Island, Alaska that was excavated in 1931 and is now curated at the Smithsonian Institution (Ousley et al., 2005; Schmidt, 2006). The Carlin and Kodiak Island samples are assumed to be individuals of Chinese ancestry. Although the Carlin sample is small, it is of historical value, with the potential to provide important insight into the origins and migration of Chinese immigrants to North America. Biological variability is not well known in early Chinese-American populations; therefore, this study also adds significant knowledge to Chinese cranial variability in a migratory context.
Two separate craniometric analyses were conducted to (1) assess levels of population history and hierarchical structure that will test whether these individuals are biologically distinct Chinese; and (2) test hypotheses of regional East Asian variation and geographic origin for the Chinese immigrants. Will the analysis of craniofacial variability allow us to pinpoint the biological origin of these Chinese immigrants? Given that the historical record suggests a South China biological and port of origin for most Chinese emigrating to North America, our analysis will attempt to substantiate these claims using biological data. Further, will Chinese samples be distinct enough to allow for such an interpretation? That is, using known samples covering the extensive geography of China, will they be distinct enough to make interpretations of birthplace for the Chinese immigrant sample?
To test for population history and get a better understanding of regional variation, the immigrant sample (pooled Carlin and Kodiak Island samples) was compared to a global dataset of diverse populations (Howells, 1973). If these individuals are indeed Chinese according to historical records, we hypothesize that the immigrant group will cluster closest phenotypically to other known East Asian groups. Based on the assumption that these individuals in fact cluster closest to known East Asian skulls, the second analysis consisted of a within-group component to better understand the biological variability of various Chinese groups and to discern a possible geographic origin for the immigrant groups. If the historical record is accepted, then our analysis should reflect a closer genetic affinity with indigenous south China populations. That is, the North American Chinese group should be similar craniometrically to groups sampled from the Canton region and Hong Kong of southern China.
Table 1 summarizes the sample names, location, temporal period, references, and sample size for each cranial series used in this study. The entire craniometric dataset consists of 1328 male crania. In order to investigate the population origins and further variation within the Chinese immigrant community, craniometric analyses were conducted on two proposed Chinese immigrant groups and comparative data taken from Howells (1973, 1989), Brown (1999), and the University of Michigan Museum of Anthropology (UMMA) craniofacial dataset (Brace et al., 2001). Importantly, Brown (1999) provides modern Chinese samples for locations in northern (Shanxi) and southern (Guangdong) China. Five additional cranial series were obtained from the UMMA dataset: Canton region, Guangxi Province, and Hong Kong of southern China; Chengdu, Sichuan Province, a Neolithic sample from Xi’an, central China, and a northern China sample from Tianjin.
Standard craniometric measurements were taken by the first author as outlined by Buikstra and Ubelaker (1994) for both Chinese immigrant samples. For craniometric analysis, we used the following 19 linear measurements: maximum cranial length, maximum cranial breadth, bizygomatic diameter, basion–bregma height, cranial base length, basion–prosthion length, maxilloalveolar breadth, maxilloalveolar length, biauricular breadth, upper facial height, nasal height, nasal breadth, orbital breadth, orbital height, biorbital breadth, frontal chord, parietal chord, occipital chord, and mastoid length (Table 2).
The Carlin Chinese immigrant cemetery consists of a small sample of 13 individuals (Owsley et al., 1997; Schmidt, 2006). These individuals were studied extensively using osteometric and paleopathological methods (Schmidt, 2006). The cemetery dates from 1885 to 1923 according to historical records and artifacts recovered with the burials (Owsley et al., 1997; Chung et al., 2005; Schmidt, 2006). These skeletal remains are currently being stored at the Department of Anthropology, University of Nevada, Las Vegas. To increase the Chinese immigrant sample size, craniometric measurements were taken for a sample of 62 individuals from the National Museum of Natural History (NMNH) in Washington, DC.
According to museum archives, the NMNH sample was excavated by an amateur archaeologist in 1931 from the site of the Karluk Fish Cannery on Kodiak Island, Alaska. The remains were then sent to Dr Ales Hrdlicka, curator of Physical Anthropology at the time (Jones, 1931; Ousley et al., 2005). The amateur archaeologist assumed these individuals to be Chinese based on the presence of a ‘pig tail’ on one of the men (Jones, 1931). Hrdlicka never tested these assumptions. Ousley et al. (2005) confirmed their biological ancestry by applying a discriminant function analysis on the Karluk sample and compared them to other known East Asian groups (Chinese and Japanese) and Native Alaskan samples. These authors found the majority of Kodiak Island individuals (95%) clustered closest to other Chinese and Japanese rather than Native Alaskan individuals. No further historical or archaeological information was available for the Kodiak Island Chinese. All Chinese immigrant individuals in the present study were assigned male sex based on both cranial and pelvic diagnostic features (Buikstra and Ubelaker, 1994; Owsley et al., 1997; Schmidt, 2006). Therefore, only male cranial data are used in the following analyses.
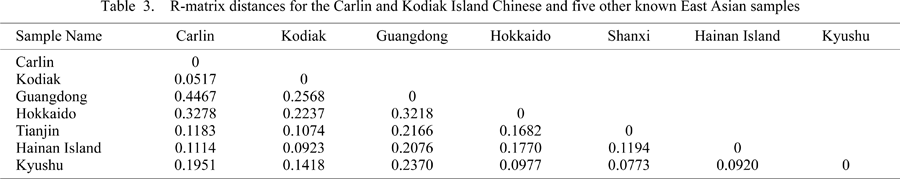
The Carlin and Kodiak samples were aggregated into a single population entitled ‘Chinese Immigrant’. This was done in order to increase the Chinese immigrant sample size. The Carlin Chinese sample consisted of only nine complete crania for use in craniometric analysis. The Kodiak Island sample was significantly larger (n = 58). We tested the biological relationship between these samples using univariate and multivariate statistics. The univariate test (independent samples t-test) showed few significant differences for several craniometric variables (data not shown). Only size measurements, such as maximum cranial breadth and maximum cranial length showed any significant differences (P < 0.05). We further subjected the Carlin and Kodiak Island samples to an R-matrix analysis and a discriminant function analysis to assess overall phenotypic difference (Figure 1, Table 3). Minimal phenotypic differentiation was observed for the two immigrant groups. Due to this observation, in combination with the historical and archaeological data, we felt comfortable combining the samples into an aggregate Chinese immigrant sample.
 View Details | Figure 1. Canonical discriminant function analysis applied to Carlin and Kodiak Island Chinese using 19 craniometric variables. |
The raw variables were subjected to principal coordinates analysis for spatial and genetic patterning. Despite the fact that sample sizes are low, the use of averages and multivariate centroids of such samples must be of interest in perceiving general tendencies by means of classical statistics. Therefore, a principal coordinate plot can be a useful tool to test population relationships. In addition, the Chinese groups were included in a population genetic analysis (Relethford and Blangero, 1990) to better understand the potential impact of gene flow or isolation after migration from China. As the genealogical histories of these individuals are unknown, a population genetic approach may explain a potential admixture event after settlement in North America.
We employed the R-matrix method on the craniometric data, and generated principal coordinates. The R-matrix method was originally proposed by Harpending and Jenkins (1973) for allele/haplotype frequency data, and was further expanded to quantitative traits by Williams-Blangero and Blangero (1989) and Relethford and Blangero (1990). Given data on means and an estimate of average heritability, an R-matrix can be estimated from quantitative traits (Relethford, 2007). An R-matrix provides estimates of genetic similarities and distances within and among populations relative to the contemporary means of allele frequencies in a region (Relethford and Harpending, 1994). Recently, the R-matrix/Relethford–Blangero method has been applied to quantitative morphological traits to search population relationships and/or population structure by many researchers (Relethford, 1994; Relethford and Harpending, 1994; Powell and Neves, 1999; Steadman, 2001; González-José et al., 2001, 2005; Roseman and Weaver, 2004; Stojanowski, 2004, 2005; Hanihara and Ishida, 2005, 2009; Hanihara, 2008; Ishida et al., 2009). Continuing this body of literature, these methods will be applied to the present study.
Because phenotypic traits are not completely under genetic control, the R-matrix method makes assessments based on estimates of the average heritability of phenotypic traits. Various studies have published the heritability estimates of craniometric traits (Sjøvold, 1984; Devor, 1987; Sparks and Jantz, 2002). According to Relethford and Harpending (1994), heritabilities for craniometric traits are fairly stable across populations, and an average estimation of h2 = 0.55 can be used (Devor, 1987). More recently, Carson (2006) published narrow-sense heritability estimated for 33 craniometric dimensions using a maximum likelihood variance components method on a skeletal sample of pedigreed individuals from Hallastatt, Austria. In that study, she found low heritability estimates of most bilateral breadth measurements, while cranial length and height dimensions have heritability values ranging between 0.102 and 0.729 (Carson, 2006). Relethford and Blangero (1990) concluded that as long as the narrow-sense heritability for a traits was greater than 0.2, the relative pattern of genetic distances between populations did not change significantly, although the absolute genetic distances will change, given different heritabilities. Thus, in this study, we follow Relethford and others’ (Hanihara, 2008; Ishida et al., 2009) overall average heritabilities for craniometric traits of h2 = 0.55 to compute the R-matrix.
The model bound method (Relethford–Blangero) compares two different measures of variation within populations: the observed and expected levels of heterozygosity (the fraction of individuals in a population that are heterozygous for a particular locus). In general, the level of heterozygosity increases with mutation and gene flow, and decreases with genetic drift. Relethford–Blangero (1990) analysis detects deviations from an equilibrium between gene flow and genetic drift derived from observed and expected values of within-group phenotypic variance.
As such, a comparison is made between the observed and expected values of heterozygosity, which can indicate something about the level of external gene flow into populations. An assumption is that the observed and expected levels of heterozygosity will be the same across all populations in the analysis. If the observed is greater than the expected, then greater than average external gene flow is likely the cause of the excess heterozygosity. If the observed is less than the expected, then that population would appear to be more isolated and has received less gene flow. Relethford and Blangero (1990) have shown that there is a proportional relationship between heterozygosity and phenotypic variation, and as such extended the Harpending and Ward (1982) model to quantitative traits as
where Vi is the average phenotypic variance over all traits in population i, Vw is the average phenotypic variance averaged over all groups, and rii and FST are estimated from quantitative traits (Relethford, 2007). This measure should be highly informative for inferring the biological diversity of groups that have maintained extensive contact through time but have had diverging histories, possibly implementing a level of isolation, and hence genetic drift, among some of the groups under analysis.
Computation of the R-matrix requires estimates of effective population sizes. In this study, we do not estimate census population sizes, as they are unknown. Due to this lack of information, we calculate the R-matrix under the assumption of equal effective population size, which makes the transformed distance matrix similar to that of classical multidimensional methods (Relethford and Harpending, 1994). We also violate the synchronic nature of the R-matrix by including samples from different time periods. However, in the global analysis of craniometric data, only two periods are significantly different from the rest (Egypt and Anyang), which are considered ancient, but modern (Bronze/Iron Ages) in terms of their craniometry (Howells, 1973). In the within-group analysis, a greater time depth is included, using samples from the Neolithic and the Bronze Age. However, their short branch lengths (Figure 4) allow us to regard their time span as insignificant and analyze these samples as separate geographic populations within the same time period (Hanihara, 2010). All population genetic analyses were performed using Rel12, a program developed by Hideyuki Umeda at the University of Tokyo and is similar in nature to RMET, developed by John Relethford.
The computed distances from the R-matrix were constructed in a neighbor-joining (NJ) procedure to visualize population structure among samples (Saitou and Nei, 1987). The NJ method expresses the structure of groupings visually in a phylogenetic, unrooted tree, or dendrogram, and also evaluates how often a particular connection between groups has occurred among trees by repeated samples generated from bootstrapping. The NJ procedure is appropriate and can be used even for populations that have not always evolved in a hierarchical manner, such as humans who often conform to a model of isolation by distance (Kalinowski, 2009). The construction of the NJ trees were performed with Rel12 using SplitsTree (Hudson and Bryant, 2006).
The two separate analyses of the Chinese immigrant samples was limited according to the comparative dataset as standard craniometric measurements differed between them. The first, global analysis consisted of 19 craniometric variables assessed among 19 modern groups and 2 ancient groups (Egypt and Anyang). Much of the comparative cranial series used in this analysis was obtained from Howells (1973, 1989) and Brown (1999). This analysis consisted of a total of 968 crania.
Due to diverse datasets and missing data, the second, within-group analysis, was restricted to 8 Chinese groups that included diverse temporal periods (ranging from the Neolithic to the present) and 10 craniometric variables: GOL, XCB, ZYB, BBH, BNL, BPL, AUB, UFHT, NLH, NLB (see Table 2 for abbreviations). Most of these comparative samples derive from the UMMA. This analysis consisted of a total of 360 crania.
Figure 1 represents the results from the disciminant function analysis applied to the Carlin and Kodiak Chinese samples separately to understand the spatial phenotypic patterning of the immigrant groups compared to other regional populations. The plot of the first two canonical functions account for 45.4% of the variation. The Carlin and Kodiak Island Chinese cluster relatively close together and form a larger cluster of known East Asian samples. Table 3 displays the phenetic distance matrix for the Carlin and Kodiak Island Chinese and several other known Chinese and Japanese samples. As further confirmation of phenotypic similarity in the two Chinese immigrant samples, the Carlin and Kodiak distances are closer together than they are to the other East Asian samples. All further analyses aggregate the two immigrant populations into ‘Chinese Immigrant’ due to this observed phenotypic similarity.
The Chinese immigrant sample and 20 comparative craniometric samples from throughout the globe were then subjected to multivariate and model-bound analyses. Figure 2 displays results for the first two principle coordinate plots, which account for 46.6% of the total variance. As expected, the Chinese immigrants cluster closest to other known Chinese groups, falling directly in between the sample from Hainan Island (southern China) and a sample of northern Chinese (Shanxi). Figure 3 represents the distances generated from the R-matrix and applied to the NJ method to produce an unrooted dendrogram for the Chinese immigrant sample and global craniometric series. Again, the Chinese immigrant sample is clustered with a branch that includes several other East Asian groups, including northern and southern Japan. Clearly, the use of 19 craniometric measurements for the Chinese immigrant population correlates with the historical evidence of Chinese ancestry.
 View Details | Figure 2. Principal coordinate plot of the Chinese immigrant series and 20 comparative population samples based on 19 craniometric measurements. |
 View Details | Figure 3. Unrooted tree of the Chinese immigrant series and 20 comparative population samples by the neighbor-joining method applied to the distance matrix of the R matrix based on 19 craniometric measurements. |
The results of the global Relethford–Blangero analysis are presented in Table 4. As shown, the Chinese immigrant sample falls within other known East Asian groups as a function of distance from the centroid (rii). The residual variance is negative (−0.027), indicating some degree of genetic drift, but not enough to be detectable or significant.
Figure 4 represents the results from the R-matrix distances applied to the NJ method for the within-group analysis. The tree indicates the Chinese immigrant group to be intermediate between Hong Kong and the central and northern groups. The Chinese immigrant group is also more genetically distinct as indicated by the branch length of this group. Figure 5 represents the first two principal coordinates accounting for 79.4% of the within-group variance. The results of the PC analysis are similar to the NJ tree; however, the Chinese immigrant sample appears as an outlier to the other groups.
 View Details | Figure 4. Unrooted tree of the Chinese immigrant series and seven comparative population samples by the neighbor-joining method applied to the distance matrix of the R-matrix based on 10 craniometric measurements. |
 View Details | Figure 5. Principal coordinate plot for within-group Chinese diversity based on 10 craniometric measurements. |
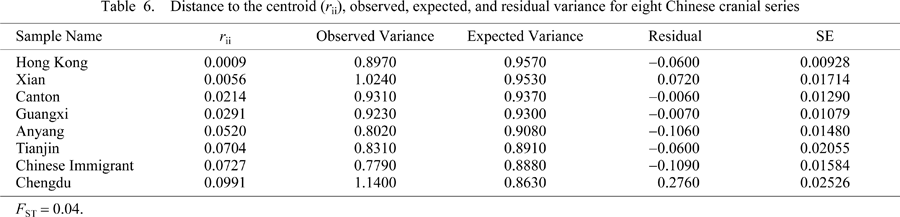
Table 5 shows biological distance values from the R-matrix. Our null hypothesis for the Chinese immigrant population to be genetically closest to other known South China groups is confirmed. As shown in the phenetic distance matrix, the immigrant sample is closest to Hong Kong, Canton, and Guangxi (located adjacent to Guangdong province). Table 6 displays the results from the Relethford–Blangero analysis for only eight Chinese samples. Most groups display a small degree of genetic drift, with the exception of the Neolithic sample and the sample from Chengdu, which is located in central China. Here, the immigrant sample displays greater than expected drift (−0.1090) compared to other Chinese populations. This could be the result of greater isolation after migrating to North America or an isolation event before emigrating.
During the late 19th and early 20th centuries, tens of thousands of Chinese immigrants settled in various cities throughout the globe, including Carlin, Nevada, and Kodiak Island, Alaska (Cassel, 2002). Those making the voyage to America as laborers came, not as slaves, but willingly through the indentured credit-ticket system (Chan, 1991). These North American Chinese immigrants came to be known as transnationalists since they were able to maintain family, socioeconomic, political, and cultural ties across international borders. Prevailing attitudes in China about overseas migration underwent significant change in the later 19th century due to a number of social, economic, and political circumstances that took place in Guangdong Province and the surrounding provinces of southern China (Hsu, 2000).
A brief survey of the domestic turmoil in Guangdong during the 19th century will help to understand why many would attempt to seek better lives in foreign lands. Cultivable land was becoming scarce, and the crops that were grown were mainly cash crops, subject to severe market price fluctuations (Hsu, 2000). Overpopulation was also becoming a problem. In 1850, a census estimated that 28 million people resided in Guangdong Province. In addition, discord in the form of internal conflicts embodied in the wars and violence between bendi, native-speaking Cantonese, and Hakka, or ‘guest peoples’ became more common. These wars scattered peoples from the countryside into cities such as Canton and Hong Kong, where they came into contact with foreigners and merchants.
Guangdong Province is located in close proximity to both Macao and Hong Kong—premier trading ports used by Western powers in the late 19th and early 20th centuries. Guangdong is currently one of the most populous areas in China and is also home to the Pearl River Delta—a series of estuaries linking local villages to trade ports and allowing access to the greater world via ports such as Canton. This region is the purported birthplace of the majority of Chinese immigrants to the United States (Chung et al., 2005) and is also the place from which the majority of Chinese immigrants set sail.
The Burlingame Treaty of 1868 established the Qing government’s policy toward Chinese emigrants in the United States, and dramatically changed centuries-old laws prohibiting migration by Chinese citizens (Cassel, 2002). This treaty enabled unrestricted migration to and from the US, and by 1880, according to census counts, there were over 100000 Chinese in the country, scattered throughout all states of the Union (Hsu, 2000). The Chinese were hired generally as laborers to work in the mines or lay railroad track across the western US. The transcontinental railroad, jointly built by the Central Pacific and Union Pacific railroad companies, employed as many as 5000 Chinese immigrants to lay tracks across northern Nevada (Chung et al., 2005).
In 1868, most individuals of Chinese descent living in the town of Carlin, Nevada worked on the transcontinental railroad. Chinese scouts were sent ahead by the railroad as early as 1860 whereupon they discovered the natural irrigation and fertile land in and around Carlin that they named ‘Chinese Gardens’ (Chung et al., 2005). The transcontinental railroad was completed in May 1869 at Promontory Summit, Utah. While over two-thirds of the 4000 men who helped complete the railroad were Chinese, at the official convocation ceremony, not a single Chinese immigrant was included in marking the occasion.
While most Chinese left the town of Carlin at this time, some chose to stay on in positions such as railroad maintenance men and construction workers for intrastate lines. Carlin, Nevada was Central Pacific’s district terminus, operating a roundhouse (for the exchange and service of locomotives), machine and car shops, a freight depot, and a refrigeration center along the transcontinental route (Chung et al., 2005). Thus while construction on the railroad ended, there were still jobs available in the town. While eventually the Carlin Chinese immigrants dwindled in number, some of these individuals died and were buried in a small, segregated, and later forgotten cemetery. It is these individuals who were buried in Carlin that formed the basis of the present study.
Craniometric variability was assessed to elucidate questions of population history surrounding two Chinese immigrant samples from North America. We first wanted to understand the general biological diversity of the samples to corroborate historical and archaeological evidence for these two groups as being Chinese in origin. Using biological distances derived from the R-matrix, a principal coordinate analysis, and a structured hierarchical tree to express relationships among the samples under analysis, we validate our original hypothesis which places the immigrant samples closest phenotypically to other known East Asian groups.
Table 3 confirms the phenotypic similarity of the Carlin and Kodiak Island Chinese. The distance matrix shows these two groups as being more similar to each other than to other Chinese and Japanese groups. Figure 1 expresses the relationship of the two Chinese immigrant samples to other regional populations. The canonical analysis clearly places both the Carlin and Kodiak Island Chinese closest to other known Chinese samples and East Asian groups. Figure 2, the principal coordinate analysis derived from the R-matrix, and Figure 3, the neighbor-joining tree, are similar to the canonical discriminant function analysis. In addition to most samples grouping geographically, there is clearly an East Asian continental branch in the NJ tree that includes the Chinese immigrant sample. These results correlate with artifacts uncovered during the Carlin excavation, which showed clear indications to Chinese ancestry (Owsley et al., 1997; Chung et al., 2005; Schmidt, 2006). Analysis of fabric and clothing styles indicated a Chinese origin. Copper Chinese coins were also recovered in several burials. In addition, five Carlin burials exhibited false queues. The queue was a particular hairstyle imposed upon the male Chinese population by the Manchu, rulers of the Qing Dynasty (1644–1911). The false queue would be worn and attached to one’s natural hair.
Another indicator of Chinese ancestry are three identification bricks recovered during the Carlin excavation and written in Chinese characters. These bricks give pertinent biographical information, such as the individual’s name, birthplace, and age at death. Furthermore, these bricks identify the year the person died and also from where they originally came from in China (village, district, and province) (Chung et al., 2005). Two bricks indicate the individual to have come from small villages in the Canton region of southern China (Tai’shan). This evidence, along with the additional archaeological material and biological analysis, all point to individuals of Chinese ancestry.
To uncover the Chinese immigrants’ place of geographic origin within the country of China, we assessed within-group variation among several known samples, including Chinese males from northern China, central China, southern China (Guangdong Province), the Canton region, Hong Kong, a Bronze Age sample, and a Neolithic sample (see Table 1). Figure 4 and Figure 5 represent the biological relationships among the Chinese groups. As shown in the NJ tree (Figure 4), the Chinese immigrant sample branches out in the middle and is located between samples from southern and northern China. The branch length for the Chinese immigrants is also longer, possibly indicating some genetic isolation for the group as a whole. The principal coordinate plot (Figure 5) also places the immigrants remote from the other samples; however, the closest samples are located geographically in southern China (Canton and Guangxi).
Due to an ambiguous placement in both the PC plot and dendrogram, we investigated the distance matrix among the Chinese samples. Table 5 represents the distances derived from the R-matrix. It is clear that the closest genetic distances to the Chinese immigrant group are the samples from southern China (Canton, Hong Kong and Guangxi). Interestingly, after Chengdu (central China, Sichuan Province), the next closest group to the immigrants is the Neolithic sample from Xi’an (in central China), while the northern Chinese sample is most distant. This could potentially indicate a biological continuity for the Chinese extending back into the Neolithic. These results are also in agreement with the finding that the majority of Chinese immigration to North America originated in the southern region of China (Chung et al., 2005).
Further, these results tentatively support the hypothesis of a north–south distinction among ethnic Chinese groups. Recent anthropometric (Du, 2004), genetic (Shi et al., 2005; Xue et al., 2006; Gao et al., 2007), and dermatoglyphic (Zhang et al., 2010) evidence indicate a split between northern and southern Chinese. Zhang et al. (2010) suggest this is most likely due to geography, as they sampled all 56 ethnic groups within China and found a general distinction between groups divided by the Yangtze River. Although overall samples used in this study are small and only a few northern Chinese samples are included (Shanxi Province and Tianjin City), a similar conclusion can be reached using craniometric variables. As indicated in both NJ trees (Figure 3, Figure 4) and principal coordinate plot (Figure 5), there is a branch separation for the northern Chinese samples from the regions of southern China.
Overall, the Relethford–Blangero analyses indicate a relatively homogeneous immigrant population (Table 4, Table 6). Given the nature and composition of an immigrant sample, we should expect to see a greater than average level of gene flow as immigrants tend to come from disparate geographic regions. This is not the case in both population genetic analyses. The results seem to indicate a relatively homogeneous group deriving from southern China. When the immigrant sample is compared with the global dataset, the residual phenotypic variance (difference between observed and expected levels of heterozygosity) is less than expected (−0.027). Interestingly, the South China sample obtained from Brown (1999) does display greater than expected phenotypic variance (0.180), as would be expected given the level of biological diversity seen in that part of the country today. In fact only the Buriat (0.298) display greater than expected levels of gene flow. Even less observed phenotypic variance is present in the Chinese immigrant sample when compared to other known Chinese populations (Table 6). Most of the populations display less than expected gene flow, or genetic isolation, with the exception of the Neolithic sample and the population from the central area of China (Chengdu). Of all the samples showing a degree of genetic drift, the Chinese immigrant samples displays the greatest level (−0.1090). This result could be explained by a recent founder effect before immigration to North America occurred.
Estimates of FST, a measure of genetic differentiation, are not unexpected in the context of regional and local craniometric studies. For the global dataset, FST is 0.15, a value similar to other studies using a large geographic craniometric dataset (Relethford, 1994). An FST estimate of 0.04 was derived for the within-group (Chinese) analysis, which is expected from a smaller geographical subset of populations. This low within-group diversity is also indicative of the insignificance of time, since it includes samples from the Neolithic and Bronze Age. These results indicate a homogeneous immigrant population originating from South China with relatively little gene flow and a degree of genetic isolation. The genetic isolation observed in the principal coordinate plot and the longer branch length in the NJ tree may have already been present in the immigrant population before it came to North America or could be the result of cultural isolation after arriving in North America.
In this study, we analyzed craniometric diversity in a Chinese immigrant sample to explore questions of population history, migration, and geographic origin. Here, the quantitative genetic analyses have confirmed earlier findings using historical and archaeological evidence. When compared to a global data set of craniometric measurements, the Chinese immigrant sample clusters closest, phenotypically, to other known East Asian groups. We further explored the within-group variability of the immigrant population by examining the diversity within a more restricted geographical space, namely groups inhabiting various regions of China. A historical reading of the Chinese immigrant experience indicates that over 90% of those individuals departed China from the Guangdong region of southern China. Here, the physical data have confirmed the historical account by showing the Chinese immigrant group is closest genetically to lineages from Canton, Hong Kong, and Guangxi, a province just north of Guangdong. By using craniometrics in an historical context, we have shown that the methodological use of craniofacial diversity is an important tool in elucidating questions of migration, population history, and population structure.
We would like to thank Dr David Hunt, Museum Specialist/Physical Anthropology Collections Management, at the National Museum of Natural History, Smithsonian Institution, USA, for access and kind permission to the Kodiak Island Chinese collection. We would also like to thank the Department of Anthropology at the University of Nevada, Las Vegas for access to the Carlin Chinese collection. This research was partly funded by the Graduate and Student Research Association at the University of Nevada, Las Vegas. The first author would also like to thank N.K. Harper for help in measuring the Carlin collection and additional multivariate analyses, and Hideyuki Umeda at the University of Tokyo for developing the computer program that conducted the population genetic analyses. We would also like to thank Tsunehiko Hanihara and two anonymous reviewers for improving the final version of this manuscript.
|