Abstract
アミロイドーシス変異アポA-Iやパーキンソン病αシヌクレインのアミロイド線維化反応に対して,核形成-自己触媒線維伸長モデルに基づく速度論的解析ならびに核形成と線維伸長速度定数の温度変動解析を行うことで,これらアミロイドタンパク質の凝集・線維化過程の熱力学的特性を明らかにした著者らの研究を紹介する.
Translated Abstract
We analyzed the kinetics and thermodynamics of amyloid fibril formation by the amyloidogenic variant of apolipoprotein A-I and α-synuclein based on the Finke-Watzky two-step model of a homogeneous nucleation followed by autocatalytic fibril growth. The results demonstrated that in apoA-I, the nucleation process is enthalpically unfavorable but entropically favorable likely because of the desolvation of hydrophobic regions in the molecule, whereas the nucleation of α-synuclein is enthalpically and entropically unfavorable. Interestingly, Parkinson’s disease-related mutation and C-terminal truncation in α-synuclein were found to decrease the enthalpic barrier for nucleation, thereby promoting autocatalytic nucleation in fibril formation.
1. はじめに
アミロイド線維は,タンパク質が規則的に重合した線維状の凝集体であり,全身の様々な臓器に沈着して機能障害を引き起こす.アミロイド線維が関わる疾患はアミロイドーシスと総称され,パーキンソン病やプリオン病,2型糖尿病,透析アミロイドーシスなど,30種類以上の疾患がある1).アミロイド線維を形成する前駆タンパク質は疾患ごとに異なるが,線維の基本構造は共通しており,βストランドが線維軸に対して垂直に折り重なったクロスβ構造をとる.アミロイド線維化はポリペプチド鎖に普遍的な性質であり,タンパク質は凝集・線維化を回避して正しくフォールドするように進化したと考えられる.天然タンパク質がどのようにして凝集・線維化を制御しているのかを理解することは,アミロイド関連疾患に対する新たな治療戦略の開拓につながると期待される.
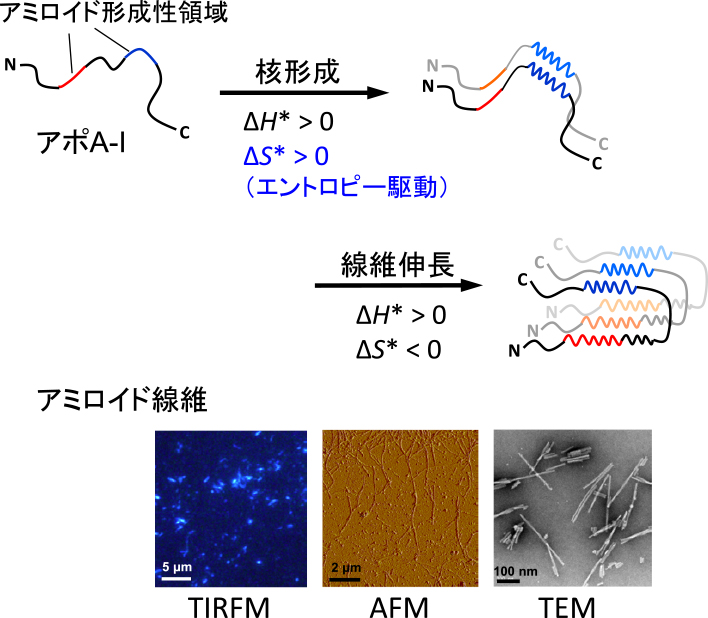
アミロイド線維形成は,円偏光二色性測定法や全反射フーリエ変換赤外分光法によるβ構造転移の検出や,線維特異的蛍光プローブであるチオフラビンT(ThT)の蛍光強度変化から追跡できる.これらの測定で得られる線維形成曲線は一般に,反応が開始するまでのラグタイムを伴ったシグモイド型曲線となる2).ラグタイムはモノマーが自発的に構造転移して不安定中間体である核が生じるのに要する時間とされ,生じた核は自己触媒的に線維へと成長する.線維化の各過程はさらに複数の微視的過程を含むが,その制御において本質的に重要な核形成と線維伸長の2つの過程に単純化したFinke-Watzkyモデルの理論式(1)は線維形成曲線をよく再現し,この式でのフィッティングから核形成と線維伸長の速度定数k1とk2を決定できる3).
|
[B
]t
=
[A
]0
-
[A
]0
k1+k2[A]0
/
k1expk1+k2[A]0t+k2[A]0
| (1) |
ここで,[A]0は時間0における全モノマー濃度,[B]tは時間tにおいて線維を形成するモノマー濃度である.この解析により,線維化の起点となる核形成と,自己触媒的な線維伸長を区別して定量評価できる.さらに,タンパク質の構造情報やAmylPred2などの凝集予測ツール4)に基づいてタンパク質に変異を導入し,核形成と線維伸長に対する影響を調べることで,凝集・線維化における各構造領域の役割が推定できる.また,反応温度やpH,タンパク質濃度等のパラメータに対する各反応速度定数の依存性を調べることで,線維化機構の速度論的・熱力学的解析が可能となる.
本稿では,全身性アミロイドーシスに関わるアポリポタンパク質A-I(アポA-I)と,パーキンソン病に関わるαシヌクレインの凝集・線維化機構について,Finke-Watzkyモデルを応用した速度論的・熱力学的解析から著者らが明らかにした最新の知見を紹介する.
2. アポA-Iアミロイド線維の形成機構
アポA-Iは,血中高密度リポタンパク質(HDL)の主要構成成分としてコレステロール恒常性維持を担う全長243残基のタンパク質であるが5),遺伝子変異や酸化修飾によりアミロイド化を起こしやすい6).アポA-IのN末端側1-83残基フラグメントは強いアミロイド線維形成性を有しており7),11種類のβ凝集予測ツールのコンセンサススコアからアミノ酸配列の凝集性を推定するAmylPred2ソフトウェア4)によって,N末端側14-22残基(LATVYVDVL)および中央部53-58残基(VTSTFS)領域が高凝集性領域と予測された.著者らは線維形成を促進するアミロイドーシス関連G26R変異を有するアポA-IのN末1-83フラグメントについて,特定の部位への蛍光標識や欠損変異体を用いた線維形成挙動の解析から,各領域の線維形成時の役割を明らかにする研究を進めてきた8).
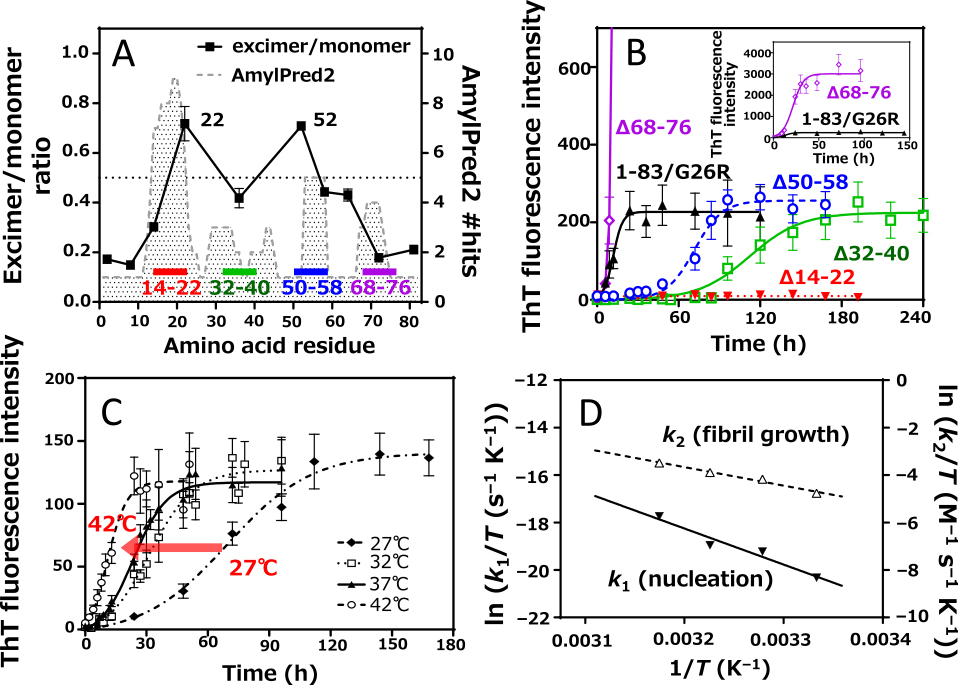
まず,様々な部位に導入したCys残基をpyrene標識し,pyrene分子同士の近接(<10 Å)により生じるexcimer蛍光を利用して各部位の凝集性を評価した結果,22および52番目残基においてpyrene excimer蛍光の著しい増大が観察され,凝集性予測の結果と一致してこの2ヶ所付近がアポA-Iアミロイド線維のコアを形成していることが明らかとなった(図1A).
次に,線維形成領域と予測される14-22残基および50-58残基領域を欠損させたΔ14-22およびΔ50-58変異体に加え,凝集傾向が低いと予測される32-40残基および電荷に富む68-76残基領域を欠損させたΔ32-40およびΔ68-76変異体を設計し,各領域の線維形成における役割を詳細に調査した.ThT蛍光経時変化から各欠損変異体の線維形成性を評価し,Finke-Watzkyモデルにより線維形成挙動を解析した結果,Δ14-22は線維形成が見られず,Δ50-58とΔ32-40では核形成と線維伸長がともに遅くなった(図1B).これは,アポA-Iの線維形成において14-22残基領域が必須の領域であり,50-58と32-40残基領域は核形成に重要な領域であることを示唆する.32-40残基領域は線維形成のコアとなる領域ではないことから,この領域は2ヶ所の線維形成領域を近接させてβシート形成を促進するヒンジ領域として働く可能性が考えられた.一方,Δ68-76はThT蛍光の著しい増大を示し,この領域がアポA-Iの線維形成に対して抑制的に働く可能性を示唆した.
さらに著者らは,アポA-I 1-83/G26Rフラグメントの核形成および線維伸長に対する熱力学的解析を行った.27~42°Cの温度範囲における線維形成挙動(図1C)から,核形成および線維伸長の各速度定数のEyring plot(図1D)を求め,両過程の活性化エンタルピーΔH*,活性化エントロピーΔS*および活性化ギブズエネルギーΔG*をそれぞれ導出した.その結果,アポA-Iの核形成はΔH*が126 kJ mol–1,ΔS*が55 J K–1 mol–1でありエンタルピー的に不利である一方でエントロピー的には有利であるが,線維伸長はΔH*が65 kJ mol–1,ΔS*が–122 J K–1 mol–1でありエンタルピー的にもエントロピー的にも不利であることが示された.一方,50-58残基領域の欠損によって核形成の活性化エントロピーが–281 J K–1 mol–1となり負の値に反転したことから,この領域がアポA-Iの核形成をエントロピー駆動的に促進することがわかった.アルツハイマー病アミロイドβペプチドにおいてもエントロピー的に有利な核形成が報告されており,その原因として凝集のコアとなる疎水性部位の脱水和が考えられている9).アポA-Iにおいても,凝集性の高い疎水性部位の脱水和によって核形成が促進されているのであろう.
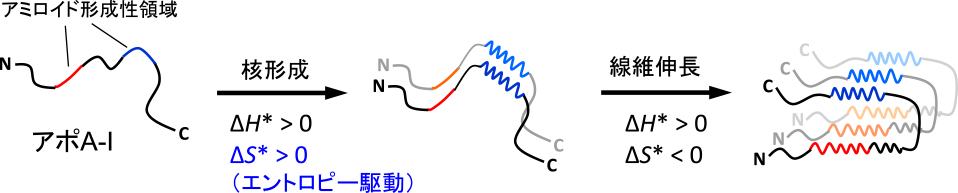
以上より,アポA-I 1-83/G26Rフラグメントのアミロイド線維では,14-22残基と50-58残基領域が線維コアを形成しており,14-22残基領域は線維形成に必須である一方,50-58残基領域は核形成をエントロピー的に駆動することがわかった(図2).
3. パーキンソン病関連αシヌクレインの凝集・線維化機構
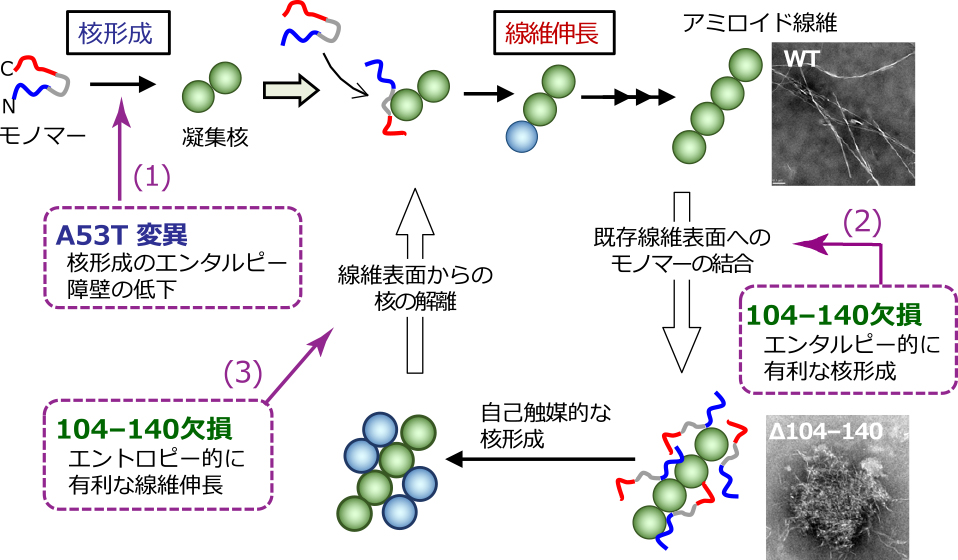
αシヌクレインは,シナプス前終末に局在する全長140残基の天然変性タンパク質であり,正電荷に富むN末領域(1-60残基),疎水性の高いNon-amlyoid β component(NAC)領域(61-95残基)および負電荷に富むC末領域(96-140残基)の3つのドメインから成る10).パーキンソン病はαシヌクレイン線維の脳内沈着を特徴とし,αシヌクレイン遺伝子の点変異や重複変異は家族性パーキンソン病の原因となる11).また,脳内に沈着したαシヌクレインの約15%はC末領域を欠損している12).ここでは,家族性変異のうちで最も頻度が高いN末A53T変異とパーキンソン病脳内に特異的なC末104-140残基領域の欠損がαシヌクレインの凝集・線維化に与える影響について,著者らが行った速度論的・熱力学的解析の結果を概説する13).
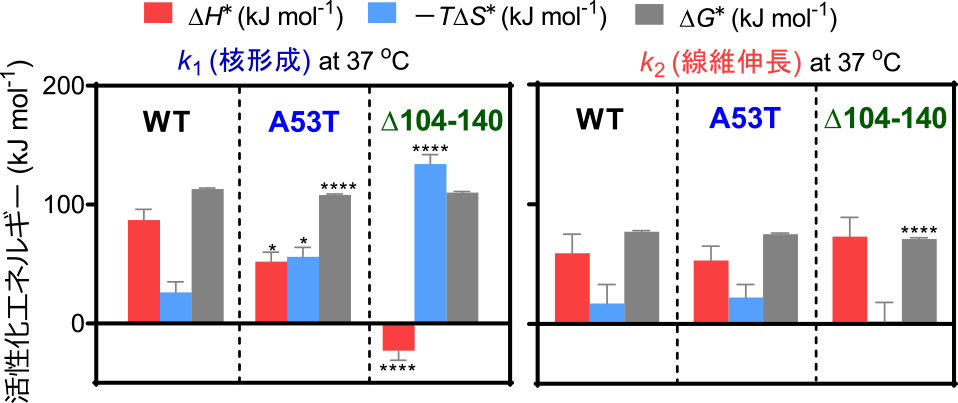
ThT蛍光経時変化測定の結果,A53T変異とC末104-140残基欠損はともに線維形成を顕著に促進した.Finke-Watzkyモデル解析から,野生型(WT)αシヌクレインではk1が1.9 × 10–3 h–1,k2が7.7 × 10–3 μM–1 h–1であり,本実験の条件であるタンパク質濃度20 μMでは核形成が線維化の律速過程であった.一方,A53T変異によってk1は1.9 × 10–2 h–1,k2は8.5 × 10–3 μM–1 h–1となり,核形成のみが顕著に加速されたが,104-140残基欠損ではk1は6.4 × 10–2 h–1,k2は2.1 × 10–2 μM–1 h–1となり,核形成と線維伸長がともに加速された.したがって,A53T変異と104-140残基欠損では線維化促進のメカニズムが異なることが示唆された.
そこで,A53T変異と104-140残基欠損による線維形成反応促進の物理化学的メカニズムを明らかにするため,Eyring plotを用いて核形成と線維伸長の熱力学パラメータを同定した(図3).WTでは,核形成と線維伸長の両過程がともにエンタルピー的にもエントロピー的にも不利であった.これはアポA-I 1-83/G26Rフラグメントの核形成がエントロピー的に有利である点と対照的である.αシヌクレインの凝集・線維化は,N末-C末領域間での分子内静電相互作用が凝集性の高いNAC領域の露出を防ぐことで抑制されていると考えられている14).そのため,WTの核形成エンタルピー障壁は,核形成に伴ったN末-C末間相互作用の切断を反映したものと考えられる.この核形成エンタルピー障壁がA53T変異体では顕著に低下することは,A53T変異が分子内N末-C末間相互作用を大きく変化させるという報告と一致する15).一方,C末104-140残基欠損によって線維化の熱力学パラメータは著しく変化し,核形成はエンタルピー的に,線維伸長はエントロピー的に有利であった.アミロイドβペプチドの凝集に関する熱力学的先行研究9)を参考にすると,104-140残基欠損体における核形成のエンタルピー的有利性は,既存線維の表面にモノマーが吸着して不安定な核形成遷移状態が安定化されること,線維伸長におけるエントロピー的有利性は線維表面で形成された核が解離して線維伸長の新たな起点となることを,それぞれ反映すると考えられた.実際,Δ104-140線維の電子顕微鏡像では短い線維が集まったクラスターが認められ(図4),104-140残基欠損による自己触媒的な二次核形成・線維伸長の結果と思われる.
以上の結果から,αシヌクレインのA53T変異はモノマーから凝集核の形成をエンタルピー的に促進して線維化の開始を早める一方,104-140残基領域の欠損はモノマーと線維の相互作用を変化させて線維化を自己触媒的に加速することが明らかとなった(図4).
4. アポA-Iとαシヌクレインの類似性
図5にアポA-Iとαシヌクレインのアミノ酸配列アライメント16)を示すが,アポA-Iの線維形成に必須の14-22残基領域はαシヌクレインの線維化を制御する36-42残基領域17)と対応していることがわかる.面白いことに,著者らが開発したアポA-I線維構造特異抗体は,この36-42残基領域に結合してαシヌクレイン線維を認識することがドッキングモデルにより示されている18).したがって,両タンパク質の線維コア領域は共通の立体構造を形成していると推察される.また,アポA-I 1-83フラグメントとαシヌクレインのいずれにおいてもC末領域の欠損によって線維化が著しく促進されたことから,負電荷に富む両タンパク質のC末領域は線維化を抑制していると考えられる.
5. おわりに
本稿では,著者らがFinke-Watzkyモデル解析で得たアポA-Iとαシヌクレインの線維化機構に関する速度論的・熱力学的知見を紹介した.このようなタンパク質の凝集・線維化過程の速度論および熱力学的特性を明らかにすることは,アミロイド線維化の分子機構解明に重要な知見を与える.
文献
- 1) Chiti, F., Dobson, C. M. (2017) Annu. Rev. Biochem. 86, 27-68. DOI: 10.1146/annurev-biochem-061516-045115.
- 2) Gade Malmos, K. et al. (2017) Amyloid 24, 1-16. DOI: 10.1080/13506129.2017.1304905.
- 3) Morris, A. M., Finke, R. G. (2009) Biophys. Chem. 140, 9-15. DOI: 10.1016/j.bpc.2008.11.003.
- 4) Tsolis, A. C. et al. (2013) PLoS One 8, e54175. DOI: 10.1371/journal.pone.0054175.
- 5) Phillips, M. C. (2013) J. Lipid Res. 54, 2034-2048. DOI: 10.1194/jlr.R034025.
- 6) Das, M., Gursky, O. (2015) Adv. Exp. Med. Biol. 855, 175-211. DOI: 10.1007/978-3-319-17344-3_8.
- 7) Adachi, E. et al. (2013) J. Biol. Chem. 288, 2848-2856. DOI: 10.1074/jbc.M112.428052.
- 8) Mizuguchi, C. et al. (2019) J. Biol. Chem. 294, 13515-13524. DOI: 10.1074/jbc.RA119.008000.
- 9) Cohen, S. I. A. et al. (2018) Nat. Chem. 10, 523-531. DOI: 10.1038/s41557-018-0023-x.
- 10) Wang, C. et al. (2016) Front. Mol. Neurosci. 9, 48. DOI: 10.3389/fnmol.2016.00048.
- 11) Fujioka, S. et al. (2014) Parkinsonism Relat. Disord. 20, S29-S34. DOI: 10.1016/S1353-8020(13)70010-5.
- 12) Baba, M. et al. (1998) Am. J. Pathol. 152, 879-884.
- 13) Ohgita, T. et al. (2022) Sci. Rep. 12, 6770. DOI: 10.1038/s41598-022-10789-6.
- 14) Stephens, A. D. et al. (2020) Nat. Commun. 11, 2820. DOI: 10.1038/s41467-020-16564-3.
- 15) Ranjan, P., Kumar, A. (2017) ACS Chem. Neurosci. 8, 2235-2246. DOI: 10.1021/acschemneuro.7b00149.
- 16) Tamura, K. et al. (2021) Mol. Biol. Evol. 38, 3022-3027. DOI: 10.1093/molbev/msab120.
- 17) Doherty, C. P. A. et al. (2020) Nat. Struct. Mol. Biol. 27, 249-259. DOI: 10.1038/s41594-020-0384-x.
- 18) Ohgita, T. et al. (2021) FEBS J. 288, 1496-1513. DOI: 10.1111/febs.15487.
Biographies
水口(深瀬)智晴(みずぐち-ふかせ ちはる)
大阪健康安全基盤研究所研究員

扇田隆司(おおぎた たかし)
京都薬科大学薬品物理化学分野助教

斎藤博幸(さいとう ひろゆき)
京都薬科大学薬品物理化学分野教授


;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/62-4ie3p01.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/62-4ie3p02.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/62-4ie3p03.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)