Abstract
An increasing number of proteins, which have neither regular secondary nor well-defined tertiary structures, have been found to be present in cells. The structure of these proteins is highly flexible and disordered under physiological (native) conditions, and they are called “intrinsically disordered” proteins (IDPs). Many of the IDPs are involved in interactions with other biomolecules such as DNA, RNA, carbohydrates, and proteins. While these IDPs are largely unstructured by themselves, marked conformational changes often occur upon binding to an interacting partner, which is known as the “coupled folding and binding mechanism”, which enable them to change the conformation to become compatible with the shape of the multiple target biomolecules. We have studied the structure and interaction of eukaryotic transcription factors Sp1 and TAF4, and found that both of them have long intrinsically disordered regions (IDRs). One of the IDRs in Sp1 exhibited homo-oligomer formation. In addition, the same region was used for the interaction with another IDR found in the TAF4 molecule. In both cases, we have not detected any significant conformational change in that region, suggesting a prominent and novel binding mode for IDPs/IDRs, which are not categorized by the well-accepted concept of the coupled folding and binding mechanism.
Significance
The “coupled folding and binding mechanism” has been suggested to be important for intrinsically disordered proteins/regions (IDPs/IDRs) in order to interact with multiple interaction partners in cells. We have studied the structure and interaction of eukaryotic transcription factors Sp1 and TAF4, and found that both of them have several IDRs. One of the IDRs in Sp1 exhibited homo-oligomer formation. The same region was also used for the heteromolecular interaction with TAF4. In both cases, no significant conformational change was detected, suggesting a prominent and novel binding mode for IDPs/IDRs.
Introduction
Almost all processes occurring in living cells, including DNA replication, gene expression, metabolism, and energy transduction, are executed by the cooperation of various proteins, sometimes in concert with RNAs and prosthetic groups. It has been suggested that the function of a particular protein is correlated with its three-dimensional structure in a one-to-one manner. For example, enzymes have a unique cavity or cleft that fits to the transition state of the compound to be catalyzed (substrate-binding pocket), and subsequent reaction proceeds efficiently by an appropriate spatial arrangement of amino acids that comprise the activity-center [1,2]. The tight and specific binding between antibody and antigen is also accomplished by suitable folding of an antibody molecule to fit the shape of the target antigen [3]. The functional form adopted by a protein under physiological conditions is called the native structure, which is unique and thermodynamically the most stable, as demonstrated by Anfinsen [4].
However, an increasing number of proteins, which have neither regular secondary nor well-defined tertiary structures, have been found to be present in cells. Furthermore, recent advances in bioinformatics and genome sequencing suggest that considerable numbers of eukaryotic proteins are predicted not to fold into the “native” structures in cells [5]. The structure of these proteins is highly flexible and disordered under physiological (native) conditions, and they are called “natively unfolded”, “intrinsically unstructured”, or “intrinsically disordered” proteins [6,7]. In some cases, most parts of a molecule adopt a well-defined tertiary structure, but have a long stretch (more than ~50 residues) of sequence that is highly flexible. Such parts of molecules are called “intrinsically disordered regions”.
Many of these intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) in proteins are involved in interactions with other biomolecules such as DNA, RNA, carbohydrates, and proteins [8,9]. While these IDPs/IDRs are largely unstructured by themselves, marked conformational changes often occur upon binding to an interacting partner, which is known as the “coupled folding and binding mechanism” [10]. This mechanism indicates that IDPs/IDRs are able to change their conformation to become compatible with the shape of the interacting partner molecules. Many IDPs/IDRs interact with not only a single but also multiple target biomolecules, and are considered to act as “hub” molecules in the interacting network composed of proteins and other biomolecules [11,12].
We have studied the structure and interaction of eukaryotic transcription factors Sp1 and TAF4, and found that both of them have long intrinsically disordered regions [13–15]. One of the IDRs in Sp1 exhibited homo-oligomer formation. In addition, the same region was used for the interaction with another IDR found in the TAF4 molecule. In both cases, we have not detected any significant conformational change in those regions, that is, the binding site of Sp1 to another Sp1 or TAF4 remains poorly structured during the process of homo- or hetero-oligomer formation. On the other hand, the binding site of TAF4 to Sp1 was also mostly disordered, but any significant conformational changes were not detected upon interaction. These observations suggest a prominent and novel binding mode for IDPs/IDRs, which are not categorized by the well-accepted concept of the coupled folding and binding mechanism.
The native structure of proteins
The native structure of a protein is stabilized by a number of interactions, including electrostatic forces, hydrogen bonds, van der Waals forces, and hydrophobic interactions [16,17]. These interactions are closely influenced by each other. For example, the strength of a Coulomb force between two are known to be dependent on a reciprocal of the square of the distance between them, and it is also reciprocally depended on the dielectric constant of the surroundings.
where q1 and q2 are the electric charge of points, ε is the dielectric constant of surroundings, and r is the distance between two charges. Equation (1) is also true for hydrogen bonds, because it is a kind of Coulomb force between functional groups induced by anisotropically distributed electrons. Importantly, most proteins, apart from membrane-integrated proteins, are surrounded by water molecules under physiological conditions, and the relative dielectric constant of water is as large as 80. This indicates that electrostatic forces and hydrogen bonds have much weaker effects on the stability of the native structure of a protein if it is exposed on the surface of a molecule. Most proteins are composed of a variety of hydrophilic and hydrophobic amino acids. As indicated by their names, hydrophilic amino acids are usually distributed on the water-accessible surface of a protein molecule, and hydrophobic amino acids are buried inside the protein to form a “hydrophobic core”. The relative dielectric constant in the “hydrophobic core” is estimated to be approximately ~10, indicating that the hydrogen bonds formed in the hydrophobic core are ~8-times stronger than those formed at the surface of a molecule, resulting in the formation of well-ordered secondary structures, which in turn induces more tight packing of hydrophobic amino acid residues. This results in a tightly packed protein molecule, with an atomic packing factor of a typical protein molecule being ~0.75, which exceeds the value of a hexagonal closest-packing structure (0.73).
Intrinsically disordered proteins/regions (IDPs/IDRs)
During the long history of evolution, not only the shape and characteristics of organisms but also the structure and function of proteins have progressed. The native structure of proteins found today is considered to be a result of the optimization of amino acid sequences under selective pressures. Therefore, every protein should fold into a unique three-dimensional structure, the native structure, in order to exert its physiological function. However, since the early 1990s, a considerable number of proteins have been found to be present in cells that do not have regular secondary nor ordered tertiary structures. Because these proteins lack unique native structures under physiological (native) conditions, they are collectively called “intrinsically disordered proteins (IDPs)”. In some proteins, not the entire molecule but a long fluctuating part of more than 50 amino acid residues is present, which are called “intrinsically disordered regions (IDRs)”.
One of the important structural features of IDPs/IDRs is that the relative amount of hydrophobic residues contained in these proteins/regions is small compared with typical globular proteins. In addition, the isoelectric point of most IDPs/IDRs markedly shifts toward acidic (pI ~ 4) or basic (pI ~ 10) regions, indicating that they are highly charged under physiological pH conditions [18]. These structural features, reduced hydrophobicity and greater charge repulsion, may prevent IDPs/IDRs from forming a well-ordered native structure, which is stabilized by the presence of a hydrophobic core and strong hydrogen bonds deep inside a compactly packed protein molecule.
Many IDPs/IDRs have been found in eukaryotic cells, especially in the nucleus. Most of them are involved in a variety of important cellular processes, including signal transduction, transcriptional activation, and cell cycle regulation. A common function underlying these processes is to interact with other biomolecules such as proteins and nucleic acids. Although IDPs/IDRs are by themselves disordered under physiological conditions, many of them acquire a well-ordered three-dimensional structure upon binding to their interaction partner molecules. This is called the “coupled folding and binding” mechanism, and it is considered as the common paradigm for the interaction of IDPs/IDRs.
We have been analyzing the homo-oligomer formation of specificity protein 1 (Sp1), one of the transcriptional activators found in eukaryotic cells. We also investigated heteromolecular interactions between Sp1 and TAF4 (TATA-box binding protein associated factor 4). We identified a region that is responsible for interaction by these proteins, and found that this region is intrinsically disordered. Moreover, we did not detect any significant conformational change in those regions upon binding, suggesting a novel interaction mechanism for IDPs/IDRs.
Transcription factors Sp1 and TAF4
The properly timed and coordinated expression of eukaryotic genes is regulated, in part, at the level of transcription initiation. The promoter-specific transcription factor Sp1 is expressed ubiquitously, and plays a primary role in regulating the expression of more than 100 genes [19]. It consists of multiple functional domains, including a C-terminal DNA-binding domain with three C2H2-type zinc fingers, and two transcriptional activation domains, A and B, which are characterized by glutamine-rich sequences (Fig. 1) [20–22]. The glutamine-rich (Q-rich) domain is one of the representative transcriptional-activation motifs found in many transcription factors and has been suggested to be involved in protein–protein interactions [23–25]. Q-rich domains have been shown to be involved in the interaction between Sp1 and different classes of nuclear proteins, such as TATA-binding protein associated factors (TAFs), which are components of the general transcription factor TFIID [26–28]. The interaction between Sp1 and TAF4, which also has four Q-rich domains in the central part of the molecule, is considered to recruit RNA polymerase II to the transcription initiation site and activate transcription.

In addition to the interaction with other proteins, the self-association of Sp1 is also important for the regulation of transcriptional activity. While binding of Sp1 to the GC-box located immediately upstream of the transcriptional start site strongly induces the expression of the encoded protein, it has also been shown that a GC-box located 1.7 kb downstream of the transcriptional start site could also act as a transcriptional enhancer. It has been considered that the Sp1 molecule that bound to the “distal” (far away from transcriptional start site) GC-box synergistically interacts with another Sp1 molecule that bound to the “proximal” (nearby the transcription site) GC-box. Furthermore, the formation of a multimeric structure by itself seems functionally important. It was indicated that the promoter activity of the transcriptionally active form of Sp1 was markedly enhanced by the addition of a DNA binding-deficient (fingerless) mutant. This synergetic effect is known as “superactivation”, and considered as a result of the interaction between Sp1 molecules via Q-rich domains [29,30].
These observations prompted us to elucidate the structure and the mechanism of interaction, which leads to the formation of homo-oligomers by Sp1 as well as the heteromolecular complex between Sp1 and TAF4.
QB domain of Sp1 is intrinsically disordered
Although there are two Q-rich domains in Sp1, we found that the QA domain did not contribute significantly to the interaction with other Sp1 molecules as well as TAF4. Our results were supported by the observation that a truncated mutant of Sp1 protein lacking the QA region was shown to possess significant transcriptional activity. Therefore, we first attempted to elucidate the conformation of the QB domain of Sp1, as well as possible structural changes upon the formation of homo-oligomers. The high-resolution solution-state NMR analysis revealed that the isolated-QB domain of Sp1 was intrinsically disordered under physiological conditions at pH 7.3 (Fig. 2A). The chemical shift dispersion along the 1H-axis was extremely narrow, and all peaks were present between 8.6 and 7.6 ppm. This is the result of a lack of strong hydrogen bonds that stabilize the secondary structures.
During the analysis of NMR spectra, we found that the peak intensity in 1H-15N HSQC spectra of the Sp1-QB domain was markedly decreased with an increase in temperature. This temperature dependency was opposite from what is observed for typical globular proteins. The peak height of Lorentzian line shape is inversely proportional to the transverse rate constant, R2, which is approximately proportional to the overall rotational correlation time, τC, of the molecule. As a protein molecule tumbles faster (smaller τC) at higher temperature, the NMR signals become sharper and their intensity should increase with temperature. Another factor that affects peak intensity of amide proton is the exchange with solvent water molecules, especially those in the experiments with “solvent suppression” pulse sequences. In this case, faster exchange rate by higher temperature is expected to result in smaller intensity. We therefore considered that the amide hydrogen atoms are exchanging easily with those in solvent water molecules, suggesting the lack of a rigid hydrophobic core that should be present in a typical globular protein.
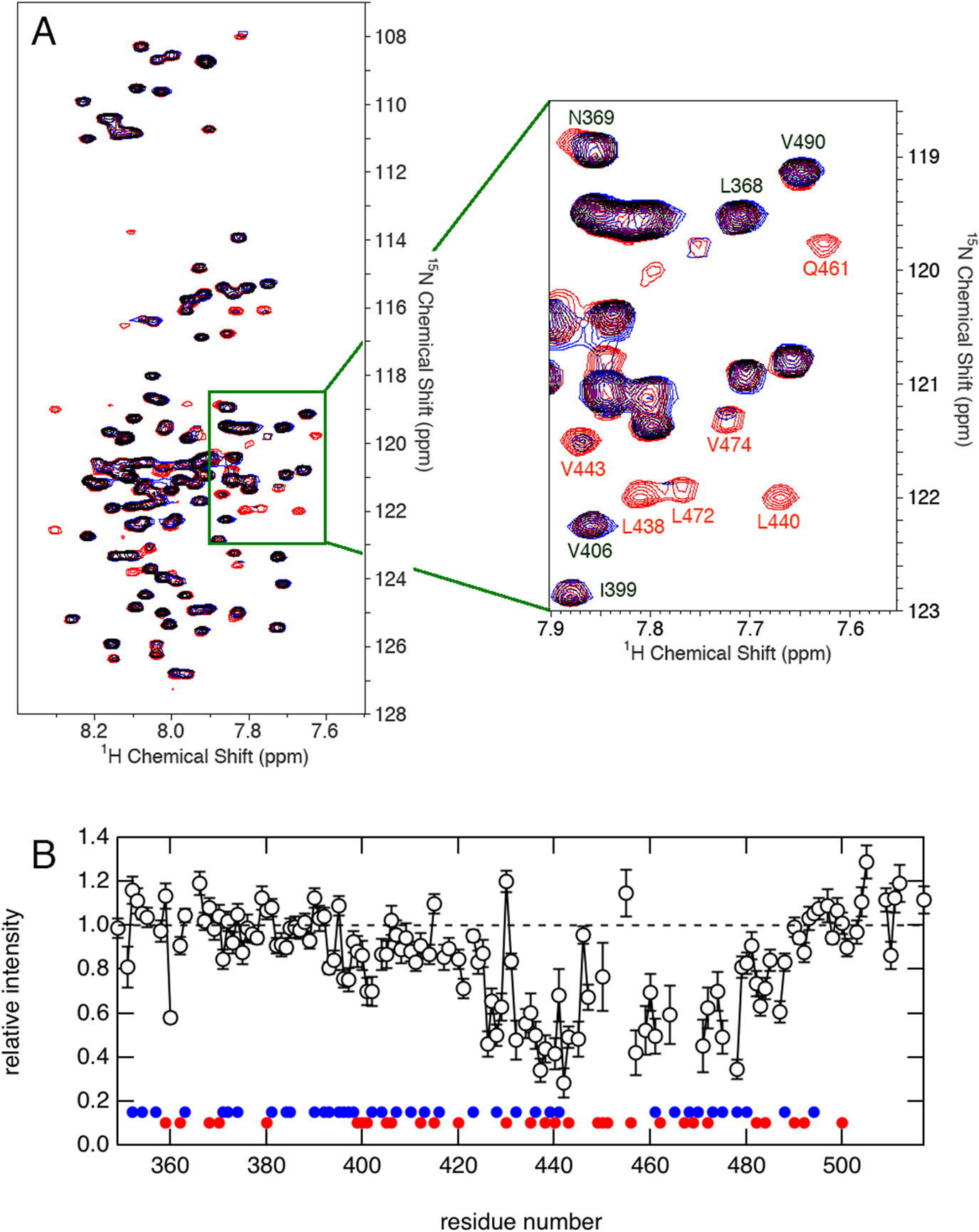
NMR peaks are sensitive indicators of changes in the local environment surrounding relevant amino acid residues. We found that a significant number of peaks in the 1H-15N HSQC spectrum of 15N-labeled QB were decreased in the presence of an excess amount of unlabeled QB domain (Fig. 2A). Note that the 1H-15N HSQC spectrum specifically detects signals from 15N spin, of which natural abundance is very low (0.37%) compared to NMR-invisible isotopic nucleus 14N (99.6%). Therefore, the presence of unlabeled QB should not affect the appearance of the 1H-15N spectrum of 15N-QB unless they interact with each other. The relative peak intensity in the presence of excess 14N-QB protein to that recorded in its absence was plotted against the residue number (Fig. 2B). It was clearly shown that the residue with a decreased intensity was located from the center to the C-terminal part of the molecule. This suggests that the interaction between isolated QB domains is site-specific, and the affected residues may represent an important binding site for the molecular interaction. A careful comparison with the amino acid type revealed that these regions are relatively rich in aliphatic residues, suggesting the involvement of hydrophobic interaction.
Although NMR results clearly indicated that particular residues were involved in homo-oligomer formation, the information of possible conformational change is missing because those residues disappeared in the spectrum. In order to elucidate this point, we measured CD spectra of QB proteins at different concentration from 50 to 300 μM (Fig. 3). All spectra perfectly overlapped, suggesting that no significant conformational change occurred at least at the secondary structural level.
Heteromolecular interaction between Sp1 and TAF4
Another important function of Sp1 is to interact with TAF4, one component of the general transcription factor that recruits RNA polymerase II in order to initiate gene transcription. We examined whether isolated QB domains from Sp1 and TAF4 interact with each other by comparing 1H-15N HSQC spectra of 15N-Sp1-QB measured in the presence or absence of an unlabeled Q-rich region derived from TAF4 (Fig. 4A). We found that selective residues located from the center to C-terminus of Sp1-QB showed a significantly decreased intensity on the addition of an excess amount of unlabeled TAF4-Q-domains (Fig. 4B). Moreover, the distribution pattern of affected residues was almost the same as that observed in Sp1-QB homo-oligomerization (Fig. 2B). This suggests that the formation of homo-oligomers by Sp1-QBs might compete with the heteromolecular interaction between Sp1-QB and TAF4-Q-domains.

The above observation clearly demonstrated that the residues located from the center to C-terminus of Sp1-QB were involved both in Sp1-homo-oligomerization and Sp1-TAF4 heteromolecular complex formation. In each case, the analysis of CD spectra suggested that no significant conformational change occurred at least at the level of the global secondary structure. However, further analyses on the structure of the resulting complex is not possible because those peaks involved in complex formation decreased in intensity and finally disappeared in the 1H-15N HSQC spectra on increasing the concentration of unlabeled partner proteins. By analyzing several sets of fragment proteins of Sp1-QB and TAF4-Q-domains, we identified the minimal fragments responsible for the interaction between Sp1 and TAF4 as the C-terminal half of Sp1-QB (QBc) and the first Q-rich domain (Q1 fragment) of TAF4. Furthermore, interaction between these fragment proteins did not result in a decrease of the peak intensity but displacement of peaks in the 1H-15N HSQC spectra in a concentration-dependent manner (Fig. 5A).

We therefore examined the 13C-chemical shift change upon interaction between Q-domains of Sp1 and TAF4 proteins. The chemical shift values of 13Cα, 13Cβ, and 13C' are known to be influenced sensitively by the ϕ and φ dihedral angles of the residue of interest, and have been used to analyze secondary structures at residual resolutions. Notably, no significant differences in 13C chemical shift values were found throughout the molecules in either protein, even for residues that showed a marked change in the chemical shift values in 1H-15N HSQC spectra (Fig. 5B). These results suggest that the interaction between Sp1 and TAF4 is not accompanied by any significant conformational changes in either protein, at least at the level of the secondary structure.
A novel interaction mode of the disordered region
One of the important structural features for these IDPs is “flexibility”, which has been suggested to enable them to access a broad conformational space to interact with a wide array of biomolecular targets. Many IDPs undergo a disorder-to-order transition to form well-defined structures upon binding to their cellular targets. This process is called coupled folding and binding, and is suggested to be a common mechanism for IDPs/IDRs to interact with their target molecules. We revealed that the C-terminal part of Sp1-QB is responsible both for the homo-oligomerization of Sp1 and for the heteromolecular interaction to form the Sp1-TAF4 complex. Furthermore, both Sp1-QB and TAF4-Q-domains were largely disordered under physiological conditions, and their conformation did not change significantly. The results of the present study suggest a prominent and novel binding mode for IDPs/IDRs, which are not categorized by the well-accepted concept of the coupled folding and binding mechanism. This novel mode of interaction might be common for the interaction between an IDP and another IDP, that is, it might be the result of two flexible IDPs mutually fitting each other. Such a phenomenon may not be a major interaction mode of IDPs, but similar examples have been reported. Sigalov et al. showed that one of the IDPs, T Cell receptor ζ chain, formed a homodimer by itself, as well as a heterodimer with the SIV Nef protein [31,32]. Another example has been found in the interaction between the C-terminal domain of Caldesmon [33]. A similar binding manner was also observed between IDR of p53 and the designed peptide [34]. These results may suggest a novel mode of interaction for IDPs that enables them to recognize many different cellular target molecules.
Conflicts of Interest
The authors declare no conflict of interest.
Author Contributions
E. H. and M. H. reviewed the studies of the interaction between intrinsically disordered proteins and wrote the manuscript.
References
- [1] Hansen, D. E. & Raines, R. T. Binding energy and enzymatic catalysis. J. Chem. Educ. 67, 483–489 (1990). DOI: 10.1021/ed067p483
- [2] Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (W. H. Freeman & Co., New York, 1999).
- [3] Davies, D. R., Padlan, E. A. & Sheriff, S. Antibody-antigen complexes. Annu. Rev. Biochem. 59, 439–473 (1990). DOI: 10.1146/annurev.bi.59.070190.002255
- [4] Anfinsen, C. B. The Formation and Stabilization of Protein Structure. Biochem. J. 128, 737–749 (1972). DOI: 10.1042/bj1280737
- [5] Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F. & Jones, D. T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 337, 635–645 (2004). DOI: 10.1016/j.jmb. 2004.02.002
- [6] Wright, P. E. & Dyson, H. J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 (1999). DOI: 10.1006/jmbi. 1999.3110
- [7] Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., et al. Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59 (2001). DOI: 10.1016/s1093-3263(00)00138-8
- [8] Dunker, A. K., Brown, C. J., Lawson, J. D., Iakoucheva, L. M. & Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 41, 6573–6582 (2002). DOI: 10.1021/bi012159
- [9] Dyson, H. J. & Wright, P. E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 (2005). DOI: 10.1038/nrm1589
- [10] Berlow, R. B., Dyson, H. J. & Wright, P. E. Functional Advantages of Dynamic Protein Disorder. FEBS Lett. 589, 2433–2440 (2015). DOI: 10.1016/j.febslet.2015.06.003
- [11] Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M. & Uversky, V. N. Flexible nets. The role of intrinsic disorder in protein interaction networks. FEBS J. 272, 5129–5148 (2005). DOI: 10.1111/j.1742-4658.2005.04948.x
- [12] Uversky, V. N., Oldfield, C. J. & Dunker, A. K. Showing Your ID: Intrinsic Disorder as an ID for Recognition, Regulation and Cell Signaling. J. Mol. Recognit. 18, 343–384 (2005). DOI: 10.1002/jmr.747
- [13] Hiramatsu, N., Hibino, E., Matsuzaki, K., Kuwahara, J. & Hoshino, M. Interaction between isolated transcriptional activation domains of Sp1 revealed by heteronuclear magnetic resonance. Protein Sci. 21, 1481–1488 (2012). DOI: 10.1002/pro.2137
- [14] Hibino, E., Inoue, R., Sugiyama, M., Kuwahara, J., Matsuzaki, K. & Hoshino, M. Interaction between intrinsically disordered regions in transcription factors Sp1 and TAF4. Protein Sci. 25, 2006–2017 (2016). DOI: 10.1002/pro.3013
- [15] Hibino, E., Inoue, R., Sugiyama, M., Kuwahara, J., Matsuzaki, K. & Hoshino, M. Identification of heteromolecular binding sites in transcription factors Sp1 and TAF4 using high-resolution nuclear magnetic resonance spectroscopy. Protein Sci. 26, 2280–2290 (2017). DOI: 10.1002/pro.3287
- [16] Makhatadze, G. I. & Privalov, P. L. Energetics of protein structure. Adv. Protein Chem. 4, 307–425 (1995). DOI: 10.1016/s0065-3233(08)60548-3
- [17] Baldwin, R. L. Energetics of protein folding. J. Mol. Biol. 371, 283–301 (2007). DOI: 10.1016/j.jmb.2007.05.078
- [18] Uversky, V. N, Gillespie, J. R. & Fink, A. L. Why Are “Natively Unfolded” Proteins Unstructured Under Physiologic Conditions? Proteins 41, 415–427 (2000). DOI: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7
- [19] Kavurma, M. M., Santiago, F. S., Bonfoco, E. & Khachigian, L. M. Sp1 phosphorylation regulates apoptosis via extracellular FasL-Fas engagement. J. Biol. Chem. 275, 4964–4971 (2001). DOI: 10.1074/jbc.M009251200
- [20] Courey, A. J. & Tjian, R. Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 55, 887–898 (1988). DOI: 10.1016/0092-8674(88)90144-4
- [21] Kadonaga, J. T., Carner, K. R., Masiarz, F. R. & Tjian, R. Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell 51, 1079–1090 (1987). DOI: 10.1016/0092-8674(87)90594-0
- [22] Elrod-Erickson, M. & Pabo, C. O. Binding studies with mutants of Zif268. J. Biol. Chem. 274, 19281–19285 (1999). DOI: 10.1074/jbc.274.27.19281
- [23] Wilkins, R. C. & Lis, J. T. DNA distortion and multimerization: novel functions of the glutamine-rich domain of GAGA factor. J. Mol. Biol. 285, 515–525 (1999). DOI: 10.1006/jmbi.1998.2356
- [24] Guo, L., Han, A., Bates, D. L., Cao, J. & Chen, L. Crystal structure of a conserved N-terminal domain of histone deacetylase 4 reveals functional insights into glutamine-rich domains. Proc. Natl. Acad. Sci. USA 104, 4297–4302 (2007). DOI: 10.1073/pnas.0608041104
- [25] Reijns, M. A., Alexander, R. D., Spiller, M. P. & Beggs, J. D. A role for Q/N-rich aggregation-prone regions in P-body localization. J. Cell Sci. 121, 2463–2472 (2008). DOI: 10.1242/jcs.024976
- [26] Hoey, T., Weinzieri, R. O. J., Gill, G., Chen, J. L., Dynlacht, B. D. & Tjian, R. Molecular cloning and functional analysis of drosophila TAF110 reveal properties expected of coactivators. Cell 72, 247–260 (1993). DOI: 10.1016/0092-8674(93)90664-c
- [27] Saluja, D., Vassallo, M. F. & Tanese, N. Distinct subdomains of human TAFII130 are required for interactions with glutamine-rich transcriptional activators. Mol. Cell. Biol. 18, 5734–5743 (1998). DOI: 10.1128/mcb.18.10.5734
- [28] Emili, A., Greenblatt, J. & Ingles, C. J. Species-specific interaction of the glutamine-rich activation domains of Sp1 with the TATA box-binding protein. Mol. Cell. Biol. 14, 1582–1593 (1994). DOI: 10.1128/mcb.14.3.1582
- [29] Courey, A. J., Holtzman, D. A., Jackson, S.P. & Tjian, R. Synergistic activation by the glutamine-rich domains of human transcription factor Sp1. Cell 59, 827–836 (1989). DOI: 10.1016/0092-8674(89)90606-5
- [30] Pascal, E. & Tjian, R. Different activation domains of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev. 5, 1646–1656 (1991). DOI: 10.1101/gad.5.9.1646
- [31] Sigalov, A. B., Zhuravleva, A. V. & Orekhov, V. Y. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 89, 419–421 (2007). DOI: 10.1016/j.biochi.2006.11.003
- [32] Sigalov, A. B., Kim, W. M., Saline, M. & Stern, L. J. The intrinsically disordered cytoplasmic domain of the T cell receptor ζ chain binds to the Nef protein of simian immunodeficiency virus without a disorder-to-order transition. Biochemistry 47, 12942–12944 (2008). DOI: 10.1021/bi801602p
- [33] Permyakov, S. E., Millett, I. S., Doniach, S., Permyakov, E. A. & Uversky, V. N. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins 53, 855–862 (2003). DOI: 10.1002/prot.10481
- [34] Kamagata, K., Mano, E., Itoh, T., Wakamoto, T., Kitahara, R., Kanbayashi, S., et al. Rational design using sequence information only produces a peptide that binds to the intrinsically disordered region of p53. Sci. Rep. 9, 8584 (2019). DOI: 10.1038/s41598-019-44688-0

