Abstract
Docosahexaenoic acid (DHA) shows more pronounced relaxation when blood vessel is contracted with prostanoid receptor agonists than other stimulants. The present study was carried out to obtain information on the mechanisms underlying prostanoid receptor-selective relaxant action of DHA, particularly focusing on the possible roles for K+ channels and its CYP epoxygenase (EOX) metabolites. In endothelium-denuded rat thoracic aorta, DHA (10−5 M) almost completely relaxed U46619 (a thromboxane A2 (TP) receptor agonist)-contracted muscle without substantially affecting noradrenaline (NA)-induced contraction. DHA-induced relaxation was not affected by a large conductance, calcium- and voltage-activated K+ (BK) channels inhibitor iberiotoxin (IbTX, 10−7 M) but was almost abolished by high-KCl (8×10−2 M) or 10−2 M tetraethylammonium (TEA) which non-selectively inhibits K+ channel activity. DHA also prominently relaxed U46619-contracted aorta even in the presence of CYP inhibitors (SKF525A or miconazole, each at 10−5 M). However, in the presence of these CYP inhibitors, the relaxant action of DHA was not affected by 10−2 M TEA. In supporting a significant role for CYP EOX metabolites in the blood vessel relaxation to DHA, 16,17-epoxy docosapentaenoic acid (16,17-EpDPE), but not 19,20-EpDPE, showed a potent relaxation in U46619-contracted aorta, and this action was significantly attenuated by 10−2 M TEA. The present findings suggest that the relaxant action of DHA shown in the rat aorta contracted through the stimulation with TP receptor is generated by DHA itself and its CYP EOX metabolites. The relaxant effect of DHA metabolites seems to be partly triggered by the activation of K+ channels though the role for BK channel is insignificant.
Intake of fish or fish oil as a supplement is known to protect against cardiovascular disorders including coronary heart diseases,1–3) atherosclerosis4,5) and stroke.6) Epidemiological studies have also indicated dietary fish oil has a blood pressure-lowering effect in hypertensive patients though this effect is weak or not observed in normotensive individuals.7–9) These cardiovascular-protective effects of fish oil are probably attributed to docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), the major n-3 polyunsaturated fatty acids (PUFAs) contained in fish oil, which show anti-inflammatory effects, reduction of platelet aggregation and of plasma triglycerides,10,11) and so on. Of these two n-3 PUFAs, DHA may be superior to EPA as a cardiovascular protector since DHA produces more favorable improving effects on abnormal lipid profile,12) thrombosis risk13) and ambulatory blood pressure.14)
Although it remains poorly understood how DHA exerts its beneficial actions on the cardiovascular systems, its cardiovascular-protective effects may be partly attributed to blood vessel relaxant effects. As to this issue, we previously found out DHA more strongly relaxes the blood vessels contracted through the stimulation with thromboxane A2 (TXA2) receptor (TP receptor) than α1-adrenoceptor in the aortae of guinea-pig15) and rat.16) TXA2 is a powerful vasoconstrictor and is suggested to play a causative role in the pathogenesis of various cardiovascular disorders including hypertension.17–19) Furthermore, its production is elevated through the stimulation with angiotensin II (Ang II) in hypertension.20) By contrast, several studies have suggested marginal roles for TXA2/TP receptors in the blood pressure regulation.21–23) Nevertheless, to reveal the mechanisms by which DHA more selectively relaxes the blood vessels contracted with TP receptor agonists may lead to better understanding of possible cardio-protective effects of DHA.
On the other hand, recent studies have suggested significant roles for DHA metabolites in the relaxant regulation of blood vessels by this n-3 PUFA. Such DHA metabolites include epoxydocosapentaenoic acid (EpDPE) regioisomers (4,5-, 7,8-, 10,11-, 13,14-, 16,17-, and 19,20-EpDPE), which are enzymatically produced by CYP-dependent epoxygenase (EOX).24) In addition, the activation of large conductance, calcium- and voltage-activated K+ (BK, MaxiK) channel has been suggested by DHA25) or its CYP EOX metabolites.26–28)
The present study was thus carried out to know whether the relaxant effect of DHA exerted on the blood vessels contracted through the stimulation with TP receptor involves activation of K+ channel with simultaneously paying attention to the possible roles for CYP EOX metabolites of DHA.
MATERIALS AND METHODS
AnimalsMale Wistar rats (8–9 weeks old, weighing 180–230 g, Sankyo Labo Service, Tokyo, Japan) were housed under controlled conditions (temperature 21–22°C, relative air humidity 50±5%, fixed 12 h-light (08 : 00 to 20 : 00)/12 h-dark cycle). Food and water were available ad libitum to all animals. This study was conducted in accordance with the Guideline for the Care and Use of Laboratory Animals adopted by the Committee on the Care and Use of Laboratory Animals of Toho University School of Pharmaceutical Sci-ences (accredited by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan).
Preparation of Rat Thoracic Aortic RingsWistar rats were anesthetized with pentobarbital sodium (30 mg/kg, intraperitoneally (i.p.)). Following disappearance of the corneal reflex, rats were killed by decapitation. A section of the thoracic aorta from between the aortic arch and diaphragm was isolated and placed in normal Tyrode’s solution (mM): NaCl, 158.3; KCl, 4.0; CaCl2, 2.0; MgCl2, 1.05; NaH2PO4, 0.42; NaHCO3, 10.0 and glucose, 5.6. The aorta was cleaned of loosely adhering fat and connective tissues and cut into ring segments of approximately 2 mm in length.
In this study, endothelium-denuded preparations were used to examine the relaxant effects of DHA to focus our attention on its smooth muscle-direct effects. This is because we have recently found out DHA-induced relaxation in rat aorta contracted with U46619 is independent of the presence of endothelium and is produced through smooth muscle-direct pathways.16) To prepare endothelium-denuded preparations, the intimal surface of rat rings was gently rubbed with an eyebrow brush. All experiments were carried out in the presence of indomethacin (3×10−6 M) to rule out the possible contribution of endogenously-generated prostanoids.
Measurement of Tension ChangesThe aortic ring segments were mounted using stainless steel hooks (outer diameter, 150–200 µm) under an optimal resting tension of 1.0 g in a 5-mL organ bath (UC-5; UFER Medical Instrument, Kyoto, Japan) containing normal Tyrode’s solution. Normal Tyrode’s solution was continuously gassed with 95% O2–5% CO2, and kept at 35.0±1.0°C (pH=7.4). Muscle tension changes were isometrically recorded with a force-displacement transducer (T7-8-240; Orientec, Tokyo, Japan) connected to a minipolygraph (Signal Conditioner: Model MSC-2; Primetech Corp., Tokyo, Japan). Aortic tension changes were recorded with PowerLab/ML-846™ and Chart™ (Version 7.0) software (ADInstruments Japan, Tokyo, Japan). Before starting tension change experiments by using various stimulants, ring preparations were equilibrated for 60 min with bathing solution (normal Tyrode’s solution) being exchanged with a fresh solution every 20 min.
After a 60-min equilibration period, to make sure aortic preparations were capable of generating normal contractile responses, they were contracted with high-KCl (8×10−2 M) Tyrode’s solution (mM): NaCl, 82.3; KCl, 80.0; CaCl2, 2.0; MgCl2, 1.05; NaH2PO4, 0.42; NaHCO3, 10.0 and glucose, 5.6. This high-KCl-induced contraction was elicited twice; first response was recorded for 20 min and second contraction was recorded for 40 min. When the muscle tension returned to a basal tension level by replacing high-KCl solution with normal Tyrode’s solution after second high-KCl-contraction, the absence of endothelium was confirmed by the lack of relaxation to acetylcholine (ACh, 10−5 M) in the preparation pre-contracted with noradrenaline (NA, 10 −7 M). After this procedure, the bathing solution was exchanged with fresh one, and aortic ring preparations were subsequently left to be re-equilibrated for another 40 min.
Evaluation of the Relaxant Effects of DHA and Its CYP EOX MetabolitesTo investigate the relaxant effects of post-treated DHA and its CYP EOX metabolites, aortic ring preparations were pre-contracted with U46619 (5×10−9 M) to produce sustained contractions. When an adequate contraction was not achieved with 5×10−9 M, the concentration of U46619 was increased to 10−8 M. After muscle contractions reached a steady-state level, DHA or its CYP EOX metabolites (16,17-EpDPE, 19,20-EpDPE) were applied to the bath medium at a desired single concentration. As a control, their vehicles (ethanol for DHA; distilled water for metabolites) were also tested. In some experiments, DHA relaxant effects were examined in the muscle contracted with NA (10−7 M) and high-KCl (8×10−2 M). At the end of experiments, to confirm the substantial maximal relaxant response level, SQ 29548 (10−7 M) (a TP receptor antagonist) or papaverine (10−4 M) was applied.
Relaxant effect of DHA on U46619-contracted aorta was also examined in the presence of CYP inhibitors. CYP inhibitors used were: SKF525A (10−5 M)27) and miconazole (10−5 M).29) They were added to the bathing medium 60 min before addition of DHA (20 min before U46619 application).
Relaxant effects of DHA and its CYP EOX metabolites on the sustainedly-contracted aorta were expressed as percent relaxation; they were calculated by considering the tension level just before addition of DHA or its metabolites as 0% relaxation, and the basal tension level before application of vasoconstrictor stimulants (U46619, NA, high-KCl) as 100% relaxation. pIC50 Value was defined as the minus logarithm of the concentrations of DHA or its metabolites to provide 50% relaxation. When the sustained muscle tension levels attained with these vasoconstrictors were required to be shown, they were expressed as the relative contraction to the second high-KCl-induced muscle tension obtained in the beginning of experiments.
Pharmacological Evaluation of the Possible K+ Channel Molecules to Trigger the Relaxant Effects of DHA and 16,17-EpDPETo pharmacologically determine the K+ channel species to mediate the relaxant effects of DHA and 16,17-EpDPE on U46619-contracted aorta, effects of selective and non-selective K+ channel inhibitors were investigated. For this purpose, each K+ channel inhibitor was applied to the bath medium 60 min before addition of DHA/16,17-EpDPE, and its effects on the relaxant responses to these PUFAs were examined. K+ channel inhibitors used in the present study were as follows: iberiotoxin (IbTX, 10−7 M) (an inhibitor for BK channel)30); charybdotoxin (10−7 M) (ChTX: an inhibitor for BK, Kv1.2, Kv1.3 and IK channels)31–33); apamin (10−7 M) (an inhibitor for SK2 and SK3 channels)34); 4-aminopyridine (10−3 M) (an inhibitor of Kv1 family and Kv3 family)35); tetraethylammonium (TEA, 10−2 M) (a non-selective K+ channel inhibitor at this concentration)35,36); PNU-37883 A (10−6 M) (an inhibitor for ATP-sensitive K (KATP) channel).37,38)
Drugs and ChemicalsThe following drugs were used: DHA, 16,17-EpDPE, 19,20-EpDPE, 9,11-dideoxy-9α,11α-meth-anoepoxy prostaglandin F2α (U46619), [1S-[1α,2α(Z),3α,4α]-7-[3-[[2-[(phenylamino)carbonyl]hydrazino]methyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid (SQ 29548) (Cayman Chemical, MI, U.S.A.); 4-aminopyridine (4-AP), indomethacin, tetraethylammonium (TEA) chloride (Sigma-Aldrich Co., MO, U.S.A.); L-noradrenaline (NA) hydrogen tartrate monohydrate, papaverine (PPV) (Wako Pure Chemical Industries, Ltd., Osaka, Japan); iberiotoxin (IbTX), charybdotoxin (ChTX), apamin (Peptide Institute, Osaka, Japan); acetylcholine (ACh) chloride (Daiichi Sankyo, Tokyo, Japan); N-(1-adamantyl)-N′-cyclohexyl-4-morpholinecarboxamidine hydrochloride (PNU-37883 A) (Axon Medchem BV, Groningen, the Netherlands); 2-diethylaminoethyl-2,2-diphenylvalerate hydrochloride (SKF525A) (Biomol, Plymouth Meeting, PA, U.S.A.); miconazole (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan). All other chemicals used in the present study were commercially available and of reagent grade.
DHA was dissolved in pure ethanol as a stock solution at 10−2 M and diluted further with distilled water to the desired concentrations. U46619 was dissolved in 70% ethanol as a stock solution at 10−4 M. SQ 29548 was dissolved in 70% ethanol as a stock solution at 10−3 M. Indomethacin was dissolved in pure ethanol as a stock solution at 10−2 M. Final ethanol concentration in the bathing medium did not exceed 0.3%, which did not affect the vascular responsiveness. Miconazole was dissolved in pure dimethyl sulfoxide (DMSO) as a stock solution at 10−2 M. Final DMSO concentration in the bathing medium did not exceed 0.1%, which did not affect the vascular responses. NA was dissolved in ascorbic acid solution (10−3 M) to prepare solutions of 10−5–10−4 M. All other drugs were prepared as aqueous solution and diluted with distilled water.
Statistical AnalysisData are presented as mean values±S.E.M. and n refers to the number of experiments. Significance of difference between mean values was evaluated by unpaired Student’s t-test, or unpaired Student’s t-test with Welch’s correction if necessary, one-way ANOVA followed by Dunnett’s or Tukey’s multiple comparison test using GraphPad Prism™ (version 4.00; GraphPad Software, San Diego, CA, U.S.A.). A p value <0.05 was considered statistically significant.
RESULTS
Selective and Potent Relaxant Effects of DHA on U46619-Contracted AortaU46619 at 5×10−9 M elicited a sustained contraction in the isolated rat aortic rings. The contraction was 1.35±0.07 g (n=20), which corresponded to 124.9±1.2% (n=20) of high-KCl (8×10−2 M)-induced contraction. When an adequate contraction was not achieved with 5×10−9 M, the concentration of U46619 was increased to 10−8 M in some experiments.
As shown in Fig. 1A, DHA relaxed U46619 (5×10−9 M)-contracted aortic rings in a concentration-dependent fashion. Its relaxant effects vs. those of its vehicle (ethanol) were statistically significant at concentrations from 3×10−6–3×10−5 M, and pIC50 value of DHA was calculated to be 5.67±0.09 (n=4) (Fig. 1B). By contrast, DHA vehicle ethanol (0.01–0.3%) did not substantially affect U46619-induced contraction. For instance, Fig. 1Aa shows that 0.3% ethanol which was used as 3×10−5 M DHA vehicle did affect a marginal effect on the contraction to U46619 (5×10−9 M).

Although the relaxant effect of DHA 3×10−6 M was significant as shown in Fig. 1Ac, its degree varied in a wide range (37.3–94.0%) (Fig. 1B). A reason for such variation was that U46619-elicited muscle tension suddenly declined precipitously after DHA application (Fig. 1Ac). Since this kind of sudden tension decline was not generated in the preparations treated with vehicle or lower concentrations of DHA, such tension change was judged to be attributed to this concentration of DHA. However, since we did not have any rationale to explain this phenomenon, this study did not focus on this issue. Instead, we used 10−5 M DHA to investigate various inhibitors' effects on the relaxant action of this n-3 PUFA, and the tension levels at 60 min after application of DHA were employed for data analysis. This concentration (10−5 M) is a physiologically achievable level of DHA in human plasma,39) which supports the significance of this study.
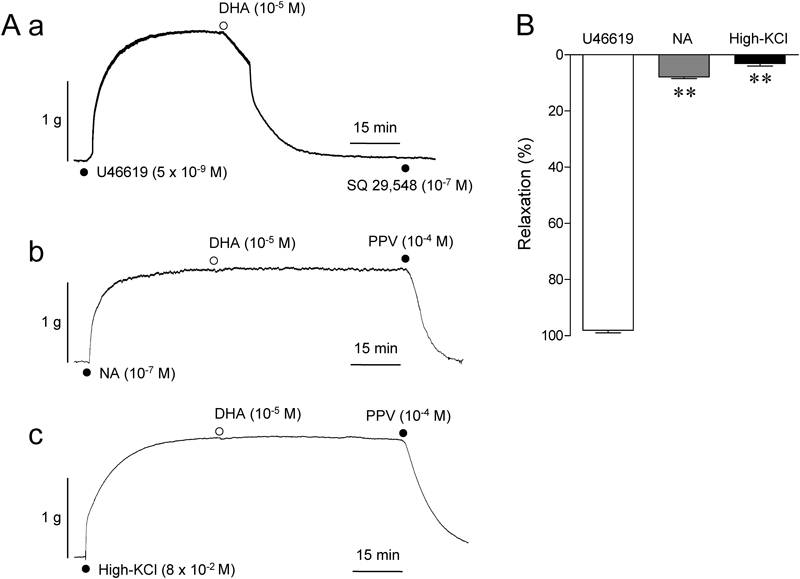
Figure 2 shows the effects of DHA (10−5 M) on the aorta sustainedly contracted by U46619, NA, and high-KCl. DHA at 10−5 M almost entirely relaxed U46619-contracted aorta (Fig. 2Aa). This was also apparent by the finding that SQ 29548 (10−7 M), a TP receptor antagonist, did not elicit any further relaxant effects. Contrastingly, DHA did not show any appreciable relaxant effects on the muscle contracted with NA (10−7 M) and high-KCl (8×10−2 M) (Figs. 2Ab, Ac, B). Because the tension level increased by U46619 (1.24±0.07 g, n=4) was significantly (p<0.01) higher than those by NA (0.97±0.05 g, n=4) and high-KCl (1.03±0.07 g, n=4), the lower amplitude of the pre-contraction level itself was ruled out as a factor to generate a selective relaxant effect of DHA on U46619-contracted aorta.
Effects of K+ Channel Inhibitors on DHA-Induced RelaxationThe finding that relaxant effect of DHA was diminished in high-KCl-contracted aorta (Fig. 2) suggested a potential role for K+ channels in the relaxant effect of DHA on U46619-contracted muscle. Therefore, in order to pharmacologically identify the responsible K+ channel candidates, relaxant effect of DHA was investigated in the presence of selective or non-selective K+ channel inhibitors. First of all, effect of IbTX, a BK channel inhibitor, was tested. However, DHA-induced relaxation was found to be unaffected by IbTX (10−7 M) (Figs. 3Ab, B). ChTX (10−7 M), another BK channel inhibitor, did not also affect DHA-induced relaxation (Figs. 3Ac, B). Similarly, apamin (10−7 M) (a SK2 and SK3 channel inhibitor) and 4-aminopyridine (4-AP, 10−3 M) (an inhibitor of Kv1 and Kv3 families) were found to be without effect (Figs. 3Ad, e, B).

By contrast, TEA (10−2 M) (a non selective K+ channel inhibitor at this concentration) (Figs. 3Af, B) and PNU-37883 A (10−6 M) (a putative KATP channel inhibitor) (Figs. 3Ag, B) profoundly suppressed DHA-induced relaxation. PNU-37883 A at 10−6 M almost abolished 3×10−7 M cromakalim (a KATP channel opener)-induced relaxation in U46619 (10−8 M)-contracted aorta from 92.5±2.1% to 5.5±0.8% (n=4, p<0.01). Cromakalim (3×10−7 M)-induced relaxation in U46619 (10−8 M)-contracted aorta was also substantially abolished by 10−2 M TEA (data not shown). Any of these K+ channel inhibitors including TEA and PNU-37883 A did not affect the aortic basal tension level (data not shown).
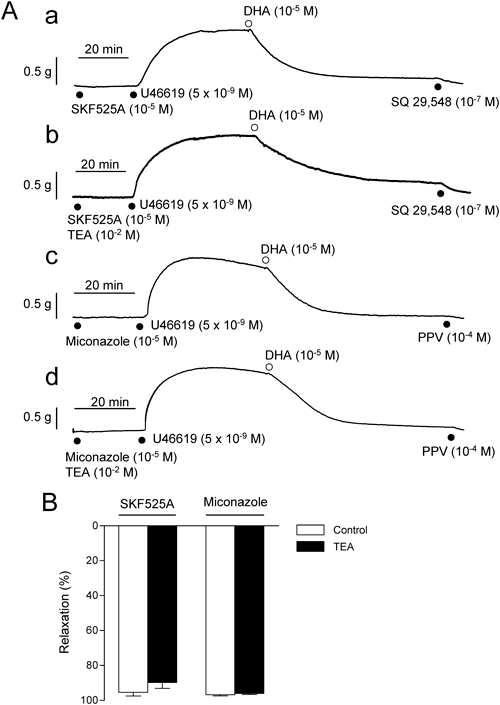
Effect of TEA on the DHA-Induced Relaxation in the Presence of CYP InhibitorsFigure 4Aa shows the DHA (10−5 M)-induced relaxant effect on U46619 (5×10−9 M)-contracted aorta in the presence of SKF525A (10−5 M). The relaxant effect of DHA was found to be preserved in the presence of this CYP inhibitor. Another CYP inhibitor miconazole (10−5 M) was also ineffective against DHA-induced relaxation (Fig. 4Ac).
On the other hand, in sharp contrast with the findings in the absence of SKF525A (Fig. 3), the preserved relaxations to DHA in the presence of SKF525A (10−5 M) and miconazole (10−5 M) were not disturbed by TEA (10−2 M) (Fig. 4Ab, Ad). Incapability of TEA to diminish DHA-induced relaxation in the presence of these CYP inhibitors are summarized in Fig. 4B. Similarly, the relaxation to DHA preserved in the presence of SKF525A (10−5 M) was not affected by PNU-37883 A (10−6 M); the relaxations were: 69.3±5.1% in the absence of PNU-37883 A and 52.9±1.3% in the presence of PNU-37883 A (n=4, p>0.05).
The presence of SKF525A (10−5 M) and miconazole (10−5 M) did not alter the inability of DHA to relax NA-contracted aorta (data not shown).
Relaxant Effects of 16,17-EpDPE and 19,20-EpDPE on U46619-Contracted Aorta and the Influences by K+ Channel InhibitorsThe above mentioned findings suggest that DHA-induced relaxation is partly attributed to K+ channels, the activation of which is triggered by CYP EOX metabolites of DHA. Therefore, in the next series of experiments, we investigated whether CYP EOX metabolites of DHA could mimic pharmacological profiles of DHA to relax U46619-contracted aorta.
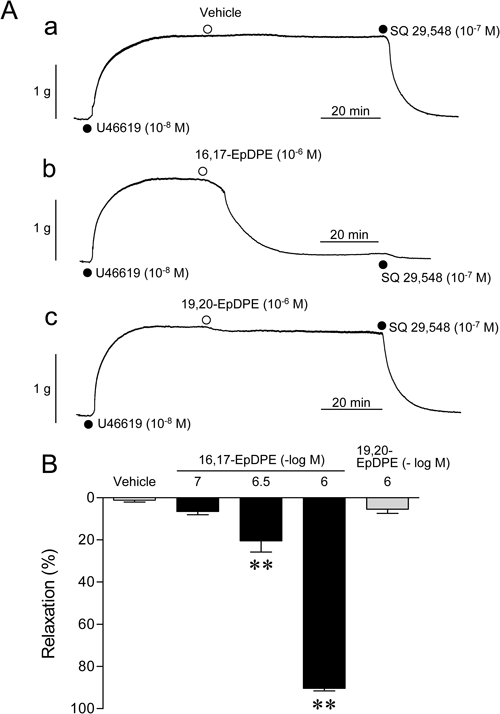
Figure 5A shows the effects of 16,17-EpDPE (Fig. 5Ab) and 19,20-EpDPE (Fig. 5Ac), CYP EOX metabolites of DHA, on U46619-contracted aorta. 16,17-EpDPE (10−6 M) almost completely relaxed the aorta contracted with U46619 whereas the same concentration of 19,20-EpDPE showed a marginal relaxant effect. The relaxant effect of 16,17-EpDPE was concentration-dependent (10−7–10−6 M). pIC50 Value of 16,17-EpDPE was 6.29±0.03 (n=3) and the maximum relaxation was 90.4±1.3% (n=3) (Fig. 5B), indicating that 16,17-EpDPE was more powerful relaxant than DHA (pIC50 value=5.67) itself by ≈4-fold.
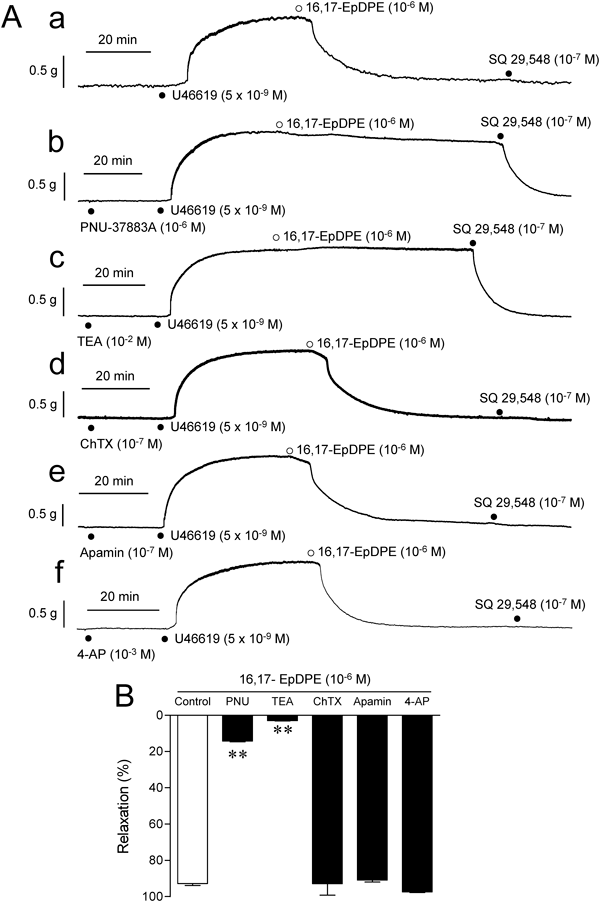
Subsequently, in order to know whether relaxant effect of 16,17-EpDPE involves activation of K+ channels, 16,17-EpDPE-induced relaxation was investigated in the absence and presence of their inhibitors and the results were shown in Fig. 6. As the relaxant effect of DHA in the absence of CYP inhibitors, the relaxation to 16,17-EpDPE was profoundly and significantly (p<0.01) prohibited by 10−2 M TEA or 10−6 M PNU-37883 A without being affected by other K+ channel inhibitors (10−7 M ChTX, 10−7 M apamin, 10−3 M 4-AP) (Fig. 6).
DISCUSSION
The present study with rat thoracic aorta suggested that DHA-induced relaxation in the artery contracted through the stimulation with TP receptor is generated by DHA itself and its CYP EOX metabolites such as 16,17-EpDPE. Roles for K+ channel seem to be significant in DHA-induced relaxation. However, plausible K+ channel activation is likely to be produced chiefly by CYP EOX metabolites of DHA rather than DHA itself. DHA itself seems to mainly elicit K+ channel-independent relaxation.
DHA was shown to selectively relax blood vessels contracted with TP receptor agonist in the aortae of guinea-pigs15) and rats,16) and the mesenteric artery of rats (our unpublished observation). Therefore, this pharmacological profile of DHA to selectively relax TP receptor agonist-contracted artery is common beyond animal species differences. However, details about the mechanisms by which DHA exhibits such selective blood vessel relaxation are still unknown. The semifinal goal of this study is to settle this issue. However, in this study we firstly predicted that the relaxant action of DHA might be triggered by some K+ channels, and if so, pharmacological identification of the responsible K+ channel seemed to be a first starting strategy. This presumption was based on the finding that DHA did not relax high-KCl-contracted aorta, in which potential K+ channel activation is masked because of the reduction in the gradation of K+ concentrations across plasma membrane.
In supporting our assumption, BK channel roles have been proposed in the blood vessel relaxant actions of DHA25) or its metabolites.26–28) Therefore, we firstly determined whether BK channel role is significant in the relaxant action of DHA. However, our results ruled out BK channel role since DHA relaxed U46619-contracted artery to the same degree both in the absence and presence of BK channel inhibitors IbTX and ChTX (Fig. 3). In the presence of 2×10−3 M TEA, which comparatively inhibits selectively BK channel at this concentration,35,36) DHA (10−5 M)-induced relaxation was diminished to less than 50% in n=8 experiments of total n=15 experiments. However, 2×10−3 M TEA did not show significant inhibitory effects in other n=7 experiments, and thus, averaged relaxant effect of DHA in the presence of this concentration of TEA was 49.6±9.7% (n=15). This result might be interpreted as positive evidence indicating a significant role for BK channel. However, a supposition of non-BK type of K+ channels that are susceptible to 2×10−3 M TEA seems to be more reasonable since IbTX and ChTX were found to be without effect against relaxation to DHA. In support of this speculation, our recent additional studies showed that 2×10−3 M TEA significantly diminishes cromakalim-induced relaxation in NA-contracted aorta (data not shown). This finding implies inhibitory effect of 2×10−3 M TEA on DHA-induced relaxation is attributed to its effect on non-BK type of K+ channels while substantiating insignificant role for BK channel suggested by the results with IbTX and ChTX. These findings were dispiriting since BK channel is abundantly expressed in rat aortic smooth muscle.35) Our results also ruled out substantial involvement of other Ca2+-activated K+ channels (SK and IK channels) since relaxant effect of DHA was not affected by apamin (10−7 M, a SK2 and SK3 channel inhibitor) and ChTX (10−7 M, an IK channel inhibitor) (Fig. 3). In addition, Kv1.5 channel role was also ruled out since 10−3 M 4-AP did not affect DHA action (Fig. 3).
Ineffective results with IbTX and ChTX raised a question about whether K+ channel activation certainly mediates DHA-induced relaxation. However, DHA-induced relaxation was surely inhibited by 2×10−3 M TEA. Furthermore, it was reduced overwhelmingly by 10−2 M TEA which non-selectively inhibits K+ channel activities (Fig. 3). This finding supported the adequacy of the interpretation of depressed relaxant action of DHA in high-KCl-contracted aorta. However, the K+ channel molecule to mediate DHA-induced relaxant effect was still unclear at this stage.
TEA at 10−2 M interferes with several K+ channel species, which include BK channel, KATP channel, Kv channels, and so on.35,36) Since BK channel contribution was ruled out, possible role for KATP channel was then investigated. For this purpose, glibenclamide is usually employed as a representative pharmacological tool to know the role for KATP channel in tissue responses. However, glibenclamide is reported to function as a competitive antagonist for TP receptor to diminish U46619-elicited contraction.40–42) In fact, we found out glibenclamide (10−5 M) strongly depressed U46619-elicited contraction in rat aorta (data not shown), and thus, investigation of this inhibitor on the DHA relaxant action was not practical. Other sulfonylurea derivatives (tolbutamide, tolazamide, gliclazide, glimepiride: 10−4 M for each) were also found to strongly suppress U46619-induced contraction (data not shown). We also tested nateglinide, a non-sulfonylurea type of KATP channel inhibitor. However, DHA-induced relaxation was not affected by nateglinide (3×10−5 M, 10−4 M) (data not shown). Ineffectiveness of nateglinide against DHA action might be attributed to this inhibitor profile that shows lower sensitivity to arterial smooth muscle type KATP channel.43)
By contrast, we happened to find out other type of KATP channel inhibitor, PNU-37883 A (10−6 M),37,38) strongly attenuated DHA-induced relaxation (Fig. 3). This inhibitor almost completely diminished KATP channel-mediated relaxation to cromakalim (3×10−7 M) (93% in the absence of PNU-37883 A vs. 6% in its presence) in U46619-contracted aorta. Since relaxation to cromakalim (3×10−7 M) in U46619-contracted aorta was almost abolished by 10−2 M TEA, the findings with PNU-37883 A vs. DHA/cromakalim were consistent with those with 10−2 M TEA. Taken together, these findings imply that KATP channel might be a candidate to mediate the relaxant response to DHA in U46619-contracted aorta only if PNU-37883 A exclusively inhibits KATP channel without affecting other non-KATP type of K+ channels. Previous findings that a representative KATP channel inhibitor glibenclamide behaves as a TP receptor antagonist40–42) could support this supposition.
Interestingly, as to the possible mediatory role for K+ channel, DHA-induced relaxation was not affected by 10−2 M TEA in the presence of CYP inhibitors (SKF525A and miconazole) (Fig. 4). These findings suggest that DHA is able to relax U46619-contracted aorta independently of K+ channel activation, and this action seems to be induced by DHA itself but not by its CYP metabolites. The finding also implies that the relaxant component mediated through K+ channel activation is significantly triggered by CYP metabolites of DHA.
Recently, investigations into the metabolic pathways of DHA and physiological effects of DHA metabolites on blood vessels have been intensively carried out, and resultantly, circumstances surrounding this issue have been revealed gradually. For instance, while a wide variety of bioactive metabolites are generated from a representative n-6 PUFA arachidonic acid (AA) via cyclooxygenase (COX), lipoxygenase (LOX) and CYP epoxygenase (EOX),44) bioactive metabolites of DHA are produced predominantly being mediated through CYP EOX rather than COX and LOX.45) In addition, although DHA is metabolized by LOX, the responsible enzyme(s) is in the endothelium rather than vascular smooth muscle.46) Considering these backgrounds, the most plausible metabolites of DHA concerning blood vessel action of this PUFA seem to be CYP EOX metabolites since our experiments were carried out in the presence of indomethacin to eliminate COX metabolites and using endothelium-removed preparations to rule out LOX metabolites.
With the source of DHA, six types of epoxy docosapentaenoic acids (EpDPEs) are generated via CYP EOX as potential bioactive metabolites; they are 4,5-, 7,8-, 10,11-, 13,14-, 16,17-, and 19,20-EpDPE.24) Among these DHA metabolites, 19,20-EpDPE is the only product generated by CYP1A1.24) By contrast, CYP2Cs (CYP2C9, CYP2C19) mainly produce 16,17-EpDPE and 19,20-EpDPE,24) and they are abundantly expressed in the vasculature.47) Therefore, in the present study, we evaluated the relaxant effects of 16,17-EpDPE and 19,20-EpDPE on U46619-contracted aorta to know whether CYP EOX metabolites roles are surely significant in DHA action. The results showed as DHA did, 16,17-EpDPE strongly relaxed U46619-contracted aorta. By contrast, 19,20-EpDPE did not mimic the relaxant effect of DHA (Fig. 5). Furthermore, the sensitivities of 16,17-EpDPE to K+ channel inhibitors were similar to those of DHA in the absence of CYP inhibitors; relaxant effects of 16,17-EpDPE were strongly attenuated by TEA (10−2 M) and PNU-37883 A (10−6 M) without being affected by other K+ channel inhibitors (ChTX, apamin, 4-AP) (Fig. 6). These findings suggest that CYP EOX metabolites of DHA are significantly capable of triggering K+ channel-mediated component of DHA relaxant action in rat aorta, and 16,17-EpDPE is one of such bioactive DHA metabolites.
In the presence of SKF525A and miconazole, relaxant effect of DHA was not affected by TEA (10−2 M). This finding implies that K+ channel-unrelated mechanisms are also significant in the relaxation by DHA of the rat aorta contracted with TP receptor agonists, and the mechanisms may be prompted by DHA itself independently of its metabolites. At present, we do not have experimental evidence supporting rationale for this phenomenon. However, it seems important to note that DHA did not substantially relax the aorta contracted with NA and high-KCl even in the presence of miconazole (data not shown). This indicates that the relaxant action of DHA in the presence of CYP inhibitors is still selective for TP receptor-mediated contraction, and thus, a special interaction between DHA and TP receptor-mediated events is implied to be present. One of such mechanisms may be a direct antagonism by DHA itself on TP receptor vs. TP receptor agonist.48) However, further studies are needed to confirm this hypothesis.
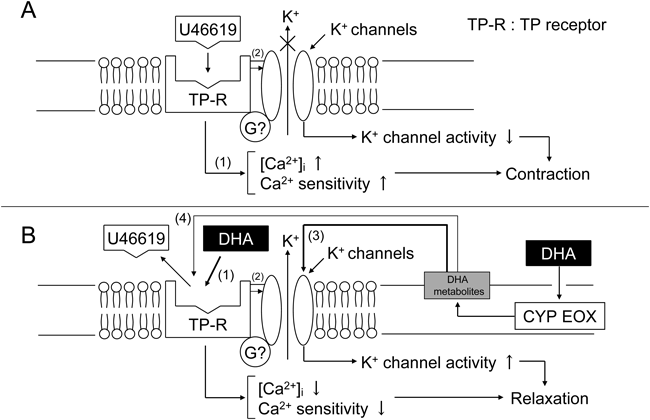
In addition, the question why K+ channels including KATP channel mediate selective relaxation by DHA/DHA metabolites of the blood vessels contracted by TP receptor agonists should be investigated in future. For instance, even if KATP channel or other K+ channels might play a substantial role in DHA-induced relaxation achieved in the artery contracted with TP receptor agonists, such K+ channel activation itself cannot be a rationale for the selectivity of DHA shown to TP receptor-mediated events. This notion could be derived from a simple observation that KATP channel activation due to cromakalim results in a strong relaxation of the arteries contracted by NA as well as by U46619 (data not shown). This is merely our supposition, but TP receptor might be physically coupled with some K+ channels, and thus these two membrane molecules may behave as a functional unit. In such situation, the first target for DHA/DHA metabolites is not K+ channel, but DHA/DHA metabolites firstly may interact with TP receptor and K+ channel activation will be secondarily achieved. Figure 7 is a figure which shows our proposed schema to explain mechanisms by which DHA/DHA metabolites produce relaxation of the rat aorta contracted with TP receptor agonists.
The concentrations of DHA (10−6–3×10−5 M) used in this study are within the practically achievable level after fish oil feeding,39) and thus, a clinical significance of this pharmacological study was suggested. In addition, our present finding provides experimental evidence to show a merit of intake of n-3 PUFA-rich fish oil as a cardiovascular protector. For instance, TXA2 is suggested to play a causative role in the pathogenesis of hypertension since SQ 29548 (a TP receptor antagonist) lowers Ang II-salt-induced blood pressure elevation.18) Therefore, possible blood pressure decrease to DHA may be partly explained by its inhibitory effects on TP receptor-activated smooth muscle events. Furthermore, other cardiovascular protective effects achievable with DHA/fish oil intake might be attributed to DHA-inhibitory effects on prostanoid receptor-mediated cardiovascular abnormalities.
In summary, our present findings indicate that DHA strongly relaxes rat aorta pre-contracted with prostanoid receptor agonists, and this action is achieved by DHA itself and its CYP EOX metabolites. 16,17-EpDPE is one of such candidates. DHA-induced aortic relaxation is partly triggered by the activation of K+ channels other than BK channel. Our present findings also imply a possible functional trans-inhibiting coupling between TP receptor and K+ channels, and DHA and its metabolites function as uncoupler of this plausible functional unit.
Acknowledgment
This study was supported in part by Grants-in-Aid for Scientific Research (C) (23590116 for YT) from the Japan Society for the Promotion of Science (JSPS), and the Science Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan (YT).
REFERENCES
- 1) Holub DJ, Holub BJ. Omega-3 fatty acids from fish oils and cardiovascular disease. Mol. Cell. Biochem., 263, 217–225 (2004).
- 2) Iso H, Kobayashi M, Ishihara J, Sasaki S, Okada K, Kita Y, Kokubo Y, Tsugane S, JPHC Study Group. Intake of fish and n3 fatty acids and risk of coronary heart disease among Japanese: the Japan Public Health Center-Based (JPHC) Study Cohort I. Circulation, 113, 195–202 (2006).
- 3) León H, Shibata MC, Sivakumaran S, Dorgan M, Chatterley T, Tsuyuki RT. Effect of fish oil on arrhythmias and mortality: systematic review. BMJ, 337 (dec23 2), a2931 (2008).
- 4) Moreno JJ, Mitjavila MT. The degree of unsaturation of dietary fatty acids and the development of atherosclerosis. J. Nutr. Biochem., 14, 182–195 (2003).
- 5) Nakamura Y, Ueno Y, Tamaki S, Kadowaki T, Okamura T, Kita Y, Miyamatsu N, Sekikawa A, Takamiya T, El-Saed A, Sutton-Tyrrell K, Ueshima H. Fish consumption and early atherosclerosis in middle-aged men. Metabolism, 56, 1060–1064 (2007).
- 6) He K, Song Y, Daviglus ML, Liu K, van Horn L, Dyer AR, Goldbourt U, Greenland P. Fish consumption and incidence of stroke: a meta-analysis of cohort studies. Stroke, 35, 1538–1542 (2004).
- 7) Appel LJ, Miller ER III, Seidler AJ, Whelton PK. Does supplementation of diet with 'fish oil' reduce blood pressure? A meta-analysis of controlled clinical trials. Arch. Intern. Med., 153, 1429–1438 (1993).
- 8) Morris MC, Sacks F, Rosner B. Does fish oil lower blood pressure? A meta-analysis of controlled trials. Circulation, 88, 523–533 (1993).
- 9) Geleijnse JM, Giltay EJ, Grobbee DE, Donders AR, Kok FJ. Blood pressure response to fish oil supplementation: metaregression analysis of randomized trials. J. Hypertens., 20, 1493–1499 (2002).
- 10) Breslow JL. n-3 fatty acids and cardiovascular disease. Am. J. Clin. Nutr., 83 (Suppl), 1477S–1482S (2006).
- 11) Lavie CJ, Milani RV, Mehra MR, Ventura HO. Omega-3 polyunsaturated fatty acids and cardiovascular diseases. J. Am. Coll. Cardiol., 54, 585–594 (2009).
- 12) Mori TA, Burke V, Puddey IB, Watts GF, OʼNeal DN, Best JD, Beilin LJ. Purified eicosapentaenoic and docosahexaenoic acids have differential effects on serum lipids and lipoproteins, LDL particle size, glucose, and insulin in mildly hyperlipidemic men. Am. J. Clin. Nutr., 71, 1085–1094 (2000).
- 13) Woodman RJ, Mori TA, Burke V, Puddey IB, Barden A, Watts GF, Beilin LJ. Effects of purified eicosapentaenoic acid and docosahexaenoic acid on platelet, fibrinolytic and vascular function in hypertensive type 2 diabetic patients. Atherosclerosis, 166, 85–93 (2003).
- 14) Mori TA, Bao DQ, Burke V, Puddey IB, Beilin LJ. Docosahexaenoic acid but not eicosapentaenoic acid lowers ambulatory blood pressure and heart rate in humans. Hypertension, 34, 253–260 (1999).
- 15) Otsuka K, Tanaka Y, Tanaka H, Koike K, Shigenobu K. Comparison of the inhibitory effects of docosahexaenoic acid (DHA) on U-46619- and phenylephrine-induced contractions in guinea-pig aorta. Biol. Pharm. Bull., 28, 1298–1300 (2005).
- 16) Sato K, Chino D, Kobayashi T, Obara K, Miyauchi S, Tanaka Y. Selective and potent inhibitory effect of docosahexaenoic acid (DHA) on U46619-induced contraction in rat aorta. J. Smooth Muscle Res., 49, 63–77 (2013).
- 17) Ogletree ML. Overview of physiological and pathophysiological effects of thromboxane A2. Fed. Proc., 46, 133–138 (1987).
- 18) Mistry M, Nasjletti A. Role of TXA2 in the pathogenesis of severe angiotensin II-salt hypertension. Adv. Prostaglandin Thromboxane Leukot. Res., 19, 207–210 (1989).
- 19) Francois H, Athirakul K, Mao L, Rockman H, Coffman TM. Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension, 43, 364–369 (2004).
- 20) Vågnes ØB, Iversen BM, Arendshorst WJ. Short-term ANG II produces renal vasoconstriction independent of TP receptor activation and TxA2/isoprostane production. Am. J. Physiol. Renal Physiol., 293, F860–F867 (2007).
- 21) De Clerck F, Beetens J, Van de Water A, Vercammen E, Janssen PA. R 68 070: thromboxane A2 synthetase inhibition and thromboxane A2/prostaglandin endoperoxide receptor blockade combined in one molecule–II. Pharmacological effects in vivo and ex vivo. Thromb. Haemost., 61, 43–49 (1989).
- 22) Patrono C, Ciabattoni G, Pinca E, Pugliese F, Castrucci G, De Salvo A, Satta MA, Peskar BA. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb. Res., 17, 317–327 (1980).
- 23) Thomas DW, Mannon RB, Mannon PJ, Latour A, Oliver JA, Hoffman M, Smithies O, Koller BH, Coffman TM. Coagulation defects and altered hemodynamic responses in mice lacking receptors for thromboxane A2. J. Clin. Invest., 102, 1994–2001 (1998).
- 24) Lucas D, Goulitquer S, Marienhagen J, Fer M, Dreano Y, Schwaneberg U, Amet Y, Corcos L. Stereoselective epoxidation of the last double bond of polyunsaturated fatty acids by human cytochromes P450. J. Lipid Res., 51, 1125–1133 (2010).
- 25) Lai LH, Wang RX, Jiang WP, Yang XJ, Song JP, Li XR, Tao G. Effects of docosahexaenoic acid on large-conductance Ca2+-activated K+ channels and voltage-dependent K+ channels in rat coronary artery smooth muscle cells. Acta Pharmacol. Sin., 30, 314–320 (2009).
- 26) Ye D, Zhang D, Oltman C, Dellsperger K, Lee HC, VanRollins M. Cytochrome P450 epoxygenase metabolites of docosahexaenoate potently dilate coronary arterioles by activating large-conductance calcium-activated potassium channels. J. Pharmacol. Exp. Ther., 303, 768–776 (2002).
- 27) Wang RX, Chai Q, Lu T, Lee HC. Activation of vascular BK channels by docosahexaenoic acid is dependent on cytochrome P450 epoxygenase activity. Cardiovasc. Res., 90, 344–352 (2011).
- 28) Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim. Biophys. Acta, 1814, 210–222 (2011).
- 29) Zhou W, Lu T, Spector AA, Lee HC. Inhibition of PGI2 signaling by miconazole in vascular smooth muscle cells. Prostaglandins Other Lipid Mediat., 80, 28–34 (2006).
- 30) Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J. Biol. Chem., 265, 11083–11090 (1990).
- 31) Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol., 45, 1227–1234 (1994).
- 32) Wallner M, Meera P, Ottolia M, Kaczorowski GJ, Latorre R, Garcia ML, Stefani E, Toro L. Characterization of and modulation by a β-subunit of a human maxi KCa channel cloned from myometrium. Receptors Channels, 3, 185–199 (1995).
- 33) Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr. Opin. Neurobiol., 8, 321–329 (1998).
- 34) Köhler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science, 273, 1709–1714 (1996).
- 35) Tanaka Y, Tang G, Takizawa K, Otsuka K, Eghbali M, Song M, Nishimaru K, Shigenobu K, Koike K, Stefani E, Toro L. Kv channels contribute to nitric oxide- and atrial natriuretic peptide-induced relaxation of a rat conduit artery. J. Pharmacol. Exp. Ther., 317, 341–354 (2006).
- 36) Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol., 268, C799–C822 (1995).
- 37) Kovalev H, Quayle JM, Kamishima T, Lodwick D. Molecular analysis of the subtype-selective inhibition of cloned KATP channels by PNU-37883 A. Br. J. Pharmacol., 141, 867–873 (2004).
- 38) Teramoto N. Pharmacological profile of U-37883 A, a channel blocker of smooth muscle-type ATP-sensitive K channels. Cardiovasc. Drug Rev., 24, 25–32 (2006).
- 39) Harper CR, Edwards MJ, DeFilippis AP, Jacobson TA. Flaxseed oil increases the plasma concentrations of cardioprotective (n-3) fatty acids in humans. J. Nutr., 136, 83–87 (2006).
- 40) Cocks TM, King SJ, Angus JA. Glibenclamide is a competitive antagonist of the thromboxane A2 receptor in dog coronary artery in vitro. Br. J. Pharmacol., 100, 375–378 (1990).
- 41) Stanke F, Cracowski JL, Chavanon O, Magne JL, Blin D, Bessard G, Devillier P. Glibenclamide inhibits thromboxane A2-induced contraction in human internal mammary artery and saphenous vein. Eur. J. Pharmacol., 341, 65–71 (1998).
- 42) Pfister SL, Pratt PE, Kurian J, Campbell WB. Glibenclamide inhibits thromboxane-mediated vasoconstriction by thromboxane receptor blockade. Vascul. Pharmacol., 40, 285–292 (2004).
- 43) Hu S, Wang S, Dunning BE. Tissue selectivity of antidiabetic agent nateglinide: study on cardiovascular and beta-cell KATP channels. J. Pharmacol. Exp. Ther., 291, 1372–1379 (1999).
- 44) Brash AR. Arachidonic acid as a bioactive molecule. J. Clin. Invest., 107, 1339–1345 (2001).
- 45) Russo GL. Dietary n-6 and n-3 polyunsaturated fatty acids: from biochemistry to clinical implications in cardiovascular prevention. Biochem. Pharmacol., 77, 937–946 (2009).
- 46) Li X, Hong S, Li PL, Zhang Y. Docosahexanoic acid-induced coronary arterial dilation: actions of 17S-hydroxy docosahexanoic acid on K+ channel activity. J. Pharmacol. Exp. Ther., 336, 891–899 (2011).
- 47) Fisslthaler B, Hinsch N, Chataigneau T, Popp R, Kiss L, Busse R, Fleming I. Nifedipine increases cytochrome P4502C expression and endothelium-derived hyperpolarizing factor-mediated responses in coronary arteries. Hypertension, 36, 270–275 (2000).
- 48) Swann PG, Venton DL, Le Breton GC. Eicosapentaenoic acid and docosahexaenoic acid are antagonists at the thromboxane A2/prostaglandin H2 receptor in human platelets. FEBS Lett., 243, 244–246 (1989).