Abstract
Thrombin-activatable fibrinolysis inhibitor (TAFI) is a carboxypeptidase B-like proenzyme biosynthesized in the liver and released into the blood circulation. Activated TAFI (TAFIa) has been implicated as an important player in maintaining the balance between blood coagulation and fibrinolysis. In the present study, regulation of TAFI (CPB2) gene expression was investigated using cultured human hepatoma HepG2 cells. HepG2 cells were treated with the phosphoinositide 3-kinase (PI3K) inhibitor LY294002, and the levels of TAFI antigen and CPB2 mRNA were measured. HepG2 cells treated with LY29400 decreased their release of TAFI antigen into the conditioned medium (CM). In parallel, there were decreased levels of CPB2 mRNA and TAFI antigen in the cells. However, CPB2 gene promoter activity was not influenced by treatment of the cells with LY294002. The half-life of the CPB2 transcript was shortened by treatment with LY294002 compared with control. The present results suggest that the PI3K inhibitor LY294002 suppresses expression of TAFI, a prothrombotic factor, by decreasing the stability of CPB2 transcripts.
Thrombin-activatable fibrinolysis inhibitor (TAFI) is a zymogen of carboxypeptidase B (CPB)-like proenzyme that is synthesized in the liver and released into circulating plasma.1) The active form of TAFI (TAFIa) is generated by cleavage at Arg92 of the proenzyme by the proteolytic activity of thrombin, plasmin, and trypsin. TAFIa exhibits CPB-like activity that specifically cleaves basic amino acids such as arginine and lysine residues located in the carboxyl (C)-termini of peptides. One of the TAFIa substrates is a partially degraded fibrin that contains a C-terminal lysine that provides a high affinity binding sites for tissue type plasminogen activator (tPA) and plasminogen in tPA-dependent plasminogen activation. Thus, TAFIa inhibits fibrinolysis by removing C-terminal lysine residues from partially degraded fibrin, thereby inhibiting development of the positive feedback mechanism in the fibrinolytic cascade.2,3) Indeed, TAFIa attenuates spontaneous fibrinolysis of batroxobin-induced fibrin deposition in rat lungs.4) TAFIa inhibitors enhance endogenous fibrinolysis, resulting in antithrombotic effects.5,6) Additionally, TAFIa has been shown to remove the C-terminal arginine residues from bradykinin as well as the anaphylatoxins C3a and C5a, thus indicating a role for the TAFI pathway in the vascular response to inflammation and the regulation of fibrinolysis.7,8)
Recent reports showed that a high level of TAFI expression in circulating plasma was a risk factor for thrombotic disorders. Thus, it was shown that elevated TAFI antigen in circulating plasma was a risk factor for premature peripheral arterial disease9) and recurrent venous thromboembolism.10) Moreover, TAFI antigen and functional protein levels in human plasma are significantly higher in the acute phase of ischemic stroke.11,12) Importantly, they have been associated with a risk of cardiovascular death in patients with coronary artery disease13) and an increased risk of first ischemic stroke.14) Hori et al.15,16) reported that elevated levels of plasma TAFI were associated with increased insulin resistance, and that treatment of 3T3-L1 adipocytes with insulin upregulated CPB2 mRNA expression in the cells. On the other hand, a different report found that plasma TAFI levels showed independent association with overall ischemic stroke, though this association was stronger for the activation peptide released from TAFI than for intact TAFI.17) Therefore, it has been suggested that TAFI plays an important role in mediating a balance between coagulation and fibrinolysis.
As noted above, TAFIa levels in circulating plasma are associated with the risk of several thrombotic vascular diseases. Thus, it is critical to understand the mechanisms regulating CPB2 gene expression. We recently showed that the CPB2 promoter lacks a consensus cAMP responsive element. Nonetheless, TAFI antigen and CPB2 mRNA levels in cultured HepG2 cells were upregulated by elevation of intracellular cAMP levels through increases in both promoter activity and the stability of CPB2 mRNA.18) Furthermore, peroxisome proliferator-activated receptor (PPAR)α agonists not only play a hypolipidemic role, but also decrease the expression of TAFI (a prothrombotic factor) through increased instability of CPB2 transcripts by mediating a function of PPARα in hepatocytes.19) On the other hand, the level of plasma TAFI (secreted by adipose tissues) might be an important factor in hypofibrinolysis in patients with insulin resistance.16) Specifically, expression of CPB2 mRNA was induced in a dose-dependent manner by insulin in 3T3-L1 adipocytes and a phosphoinositide 3-kinase (PI3K) inhibitor blocked insulin-induced CPB2 expression. These findings led us to study the involvement of the PI3K signaling pathway in the regulation of CPB2 expression in human hepatoma cells. The present work was performed to evaluate the effect of the PI3K inhibitor LY294002 on the expression and release of TAFI from cultured human hepatoma HepG2 cells.
MATERIALS AND METHODS
MaterialsLY294002, penicillin, and streptomycin were purchased from Sigma Chemical (St. Louis, MO, U.S.A.). Mouse monoclonal anti-human TAFI antibody (clone 2A16) and horseradish peroxidase (HRP)-labeled mouse anti-human TAFI antibody (10G1-HRP) were obtained from the Institute for Protein Science Co. (Nagoya, Japan). MEM and Dulbecco’s phosphate-buffered saline (−) (PBS(−)) were purchased from Nissui Pharmaceutical Co. (Tokyo, Japan). Primers for polymerase chain reaction (PCR) or real-time RT-PCR were purchased from Sigma Genosys (Tokyo, Japan). Other reagents were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan), unless otherwise indicated.
Cell CultureThe human hepatoma cell line HepG2 was obtained from the American Type Culture Collection (Rockville, MD, U.S.A.). The cells were maintained in MEM supplemented with 10% heat inactivated fetal bovine serum (FBS), 50 units/mL penicillin, 50 mg/mL streptomycin and grown in an atmosphere of 5% CO2 at 37°C. Cells were seeded at a concentration of 2–3×105 cells/mL and maintained in logarithmic growth by passing them every two or three days.
Analysis of CPB2 mRNA Expression by Real-Time Reverse Transcription (RT)-PCRTotal RNA from HepG2 cells was extracted with ISOGEN (Nippongene, Toyama, Japan) and reverse transcribed with the SuperScript II preamplification system (Invitrogen, Carlsbad, CA, U.S.A.). The sequences of the oligonucleotides used in PCR reactions were as follows: CPB2-forward, 5′-GGG TGG TCA GAA GTA CAT TAA AGA TTT-3′ (1644–1670); CPB2-reverse, 5′-CTG GGC CAT CGC CTA ATT AC-3′ (1742–1761); β-actin (ACTB)-forward, 5′-CAC TAA GTC AGG AGA TTG G-3′ (969–988); ACTB-reverse, 5′-CTC CAG GTA AGG TGT GAA C-3′ (1239–1258). The numbering of the primers is based on the numbering of the published cDNA sequences of CPB220) and ACTB.21) PCR reactions were performed using Power SYBR® Green PCR™ Master Mix (Applied Biosystems), and assayed on the ABI 7300 Detector (Applied Biosystems). The transcript number of ACTB was quantified, and each sample was normalized on the basis of ACTB content. In the case of mRNA decay assays, glyceraldehyde-6-phosphate dehydrogenase (GAPDH) cDNA was used for normalization of the amount of CPB2 mRNA. The amount of CPB2 mRNA after treatment with actinomycin D was calculated as described previously,21) assuming an 8 h half-life for GAPDH mRNA.22,23)
TAFI Antigen Levels in HepG2Confluent HepG2 cells on 24-well plates were exposed to the indicated concentrations of agents. After incubation, the cells were washed twice with PBS and lysed in lysis buffer [20 mM Tris–HCl (pH 7.4), 0.15 M NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1% Triton X-100, 1 µg/mL leupeptin, 1 µg/mL aprotinin, and 1 mM phenylmethylsulfonyl fluoride]. The lysate was centrifuged at 15000×g for 15 min, and the supernatants were subjected to a sandwich ELISA that consisted of mouse monoclonal anti-human TAFI antibody (clone 2A16) and HRP-labeled mouse anti-human TAFI antibody (10G1-HRP). TAFI antigen levels in culture medium were assayed under the same experimental conditions. TAFI protein was isolated from normal human plasma as described previously with slight modifications.4,24) Plasminogen-depleted plasma was brought to 80 mM BaCl2. After centrifugation, the supernatant, barium-adsorbed plasma (BAP), was precipitated between 35% and 70% (NH4)2SO4 saturation. TAFI in the preparation was purified by chromatography on Q-Sepharose Fast Flow, Heparin-Sepharose CL-6B, Sephacryl S-300, and plasminogen-Sepharose columns, and stored at −80°C. These chromatographic steps achieved a 12300-fold purification with a 14% recovery of TAFI activity from the BAP fraction of human plasma. The purified TAFI migrated as a single band with a molecular weight of 58 kDa on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (12% polyacrylamide) under both non-reducing and reducing conditions.25) Protein concentration was determined by the method of Bradford,26) using bovine serum albumin (BSA) as a standard. All blood samples were obtained after informed consent.
Reporter Gene AssaysHepG2 cells were transfected either with empty vector (pGL4 Basic) or with the assay plasmid for CPB2 promoter activity (pCPB2-Luc) by using TransIT transfection reagent (Mirus Corp.) according to the manufacturer’s instructions. Reporter plasmid pCPB2-Luc was constructed as described previously.19) Transfection efficiency was assessed by cotransfection with Renilla luciferase internal control plasmid (pRL-null, Promega). To investigate CPB2 promoter activity, reporter plasmid pCPB2-Luc was cotransfected with pRL-null. After a 24 h incubation, the transfected cells were treated with the indicated concentrations of LY294002 for 24 h. The detection of luciferase activity was performed with Dual-Glo™ luciferase assay system (Promega).
Statistical AnalysisData are expressed as mean±standard deviation (S.D.). Results were analyzed using Student’s t-test, and statistical significance for all comparisons was assigned at p<0.05.
RESULTS
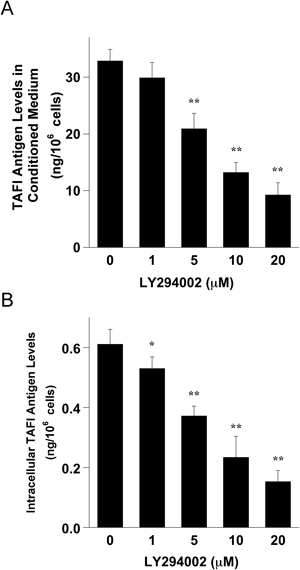
Effects of a PI3K Inhibitor on the Release of TAFI Antigen from Cultured HepG2 Cells and on Intracellular Antigen LevelsSince a recent report suggested that the PI3K signaling pathway was involved in the regulation of TAFI expression,16) we investigated the effects of the PI3K inhibitor LY294002 on the amounts of TAFI antigen released from cultured human hepatoma HepG2 cells into CM (Fig. 1A). Over a period of 48 h, 1×106 control HepG2 cells released 32.8±2.0 ng of TAFI antigen into the CM. Treatment of the cells with LY294002 decreased TAFI antigen release in a dose-dependent manner and the inhibition was significant compared with the control cells. The effects of the PI3K inhibitor on intracellular levels of TAFI antigen were further investigated. As shown in Fig. 1B, there was an apparent dose-dependent decrease in intracellular TAFI antigen levels in the LY294002-treated cells compared with the levels in control cells. Thus, both intracellular and extracellular TAFI antigen levels were reduced by treatment of the cells with PI3K inhibitor LY294002.
Decreases in the Levels of TAFI Antigen and CPB2 mRNA in HepG2 Cells Treated with LY294002We also investigated the time- and dose-dependent effects of LY294002 on TAFI antigen release from cells. There was an apparent decrease in the amount of TAFI antigen released from LY294002-treated cells into CM after 12–24 h of LY294002 treatment compared with control cells (Fig. 2A). In a dose-dependence study of LY294002 treatment, the lowest release was observed in cells treated with 20 µM LY294002 for 48 h (Fig. 2A). On the basis of the results shown in Fig. 2A, the average rates of TAFI antigen release per hour were calculated for 0 to 6 h, 6 to 12, 12–24 and 24–48 h after treatment of the cells with or without LY294002 (Fig. 2B). The average release rate of the control cells was 0.74–0.79 ng/h/106 cells for the measured periods of 6–24 h. However, a slight decrease in release rate was observed during the 24–48 h period in control cells. On the other hand, the release rates of LY294002-treated cells were in the range from 0.57 to 0.10 ng/h/106 cells during the 6–48 h period of treatment with 20 µM LY294002, and the lowest rate was observed from 24 to 48 h of treatment.
Intracellular levels of TAFI antigen and CPB2 mRNA were also determined in HepG2 cells treated with or without LY294002 (Figs. 3A, B). The antigen levels were downregulated by treatment of the cells with 1–20 µM LY294002 in a time-dependent manner and the lowest level of antigen was observed 48 h after stimulation with 20 µM LY294002 (Fig. 3A), changes similar to the average release rates of TAFI antigen from the LY294002-treated cells (Fig. 2B). CPB2 mRNA levels were also downregulated by treatment of the cells with LY294002. The lowest levels of CPB2 mRNA were found 48 h after stimulation with 20 µM LY294002 (Fig. 3B), parallelling the changes in antigen levels.
Effects of LY294002 on the Promoter Activity of the CPB2 Gene and the Stability of the CPB2 TranscriptWe asked whether the PI3K inhibitor affected CPB2 gene expression. Thus, CPB2 promoter activity in HepG2 cells was determined with a luciferase reporter construct containing the 5′-flanking region of the human CPB2 gene (Fig. 4). Although the levels of CPB2 mRNA were reduced by exposure to LY294002 in a dose-dependent manner compared with untreated control cells (Fig. 4A), CPB2 promoter activity was unaffected by LY294002 treatment (Fig. 4B). In a previous report, similar results were obtained by treatment of the cells with a combination of IL-1β and IL-6 that is known to decrease CPB2 mRNA without affecting the promoter activity of CPB2 gene expression.27) Since the downregulation of CPB2 mRNA expression by PI3K inhibition did not seem to result from a suppression of CPB2 promoter activity, the stability of CPB2 mRNA in LY294002-treated cells was determined by real-time PCR. As shown in Fig. 5, a decrease in half-life of CPB2 mRNA was observed by treatment of the cells with LY294002 compared with untreated control cells. These results sugggest that changes in the stability of CPB2 mRNA were involved in the downregulation of CPB2 mRNA by LY294002.
DISCUSSION
The present work demonstrated that treatment of cultured HepG2 cells with a PI3K inhibitor (LY294002) decreased TAFI antigen release from the cells into the CM. There was a parallel downregulation of the levels of intracellular TAFI antigen and CPB2 mRNA in PI3 kinase inhibitor-treated cells, suggesting that the LY294002-mediated decrease in TAFI antigen release was due to repression of CPB2 gene expression. The present work further investigated whether the LY294002-mediated downregulation of CPB2 gene expression was mediated by mRNA stability. Treatment of HepG2 cells with PI3K inhibitor enhanced CPB2 mRNA degradation. These results suggested that downregulation of CPB2 gene expression in LY294002-treated HepG2 cells was mediated by the regulation of CPB2 mRNA stability.
Several reports found that the 5′-flanking region of the human CPB2 gene lacked a consensus TAT A sequence and that it had several potential binding sites for transcription factors, including HNF-1α28), C/EBP29) and GR.27) Our recent work demonstrated that increased intracellular cAMP levels upregulated CPB2 mRNA expression.18) Moreover, we found that activation of the cAMP/PKA signaling pathway was involved in transcriptional and post-transcriptional upregulation of CPB2 mRNA levels. However, CPB2 promoter activity as measured with a luciferase reporter assay was unaffected by treatment of HepG2 cells with LY294002 compared with untreated control cells. The result suggested that LY294002 did not affect CPB2 transcriptional activity. Thus, the 5′-flanking region of the human CPB2 gene is not important for LY294002-induced alteration of CPB2 mRNA levels.
Our results suggested that the half-life of CPB2 mRNA in LY294002-treated cells (as determined by real-time PCR) was shortened compared to untreated controls. These results suggested that LY294002-induced downregulation of CPB2 mRNA levels was mediated by increasing the instability of CPB2 mRNA through the PI3 kinase pathway. Boffa et al. previously demonstrated that treatment of HepG2 cells with IL-1β plus IL-6 decreased CPB2 mRNA expression. This decrease was due to increased instability of CPB2 mRNA.27) Specific cis-acting elements in mRNA, such as AU-rich elements (ARE) in the 3′-untranslated region (UTR) or 3′-polyadenylate [poly(A)] tails, are involved in the regulation of the stability of mRNAs in response to various extracellular stimuli. ARE contain copies of the pentameric AUUUA sequence or the nonameric UUAUUUAUU sequence and have a high content of U residues. The destabilizing effect of AREs can be modulated via interaction of these cis sequences with A/U binding proteins that are thought to either stabilize or destabilize the target transcripts.30) A recent report indicated that cis-acting sequences in the CPB2 3′-UTR play an important role in regulating CPB2 mRNA stability. Treatment of HepG2 cells with IL-1β and IL-6 increased the instability of CPB2 mRNA through abundant formation of polyadenylated CPB2 transcripts. This result suggested the presence of cytokine-inducible instability elements in the CPB2 3′-UTR.31) The present study did not investigate the precise mechanism of LY294002-induced CPB2 mRNA instability. The destabilizing of CPB2 transcripts might be modulated via interaction of 3′-UTR sequences with A/U binding proteins that are regulated by the PI3K/Akt signaling pathway. Further work will be needed to identify PI3 kinase pathway-specific elements and the binding proteins that regulate the stability of CPB2 mRNA.
A recent report found that insulin increased the expression of CPB mRNA levels in 3T3-L1 adipocytes.16) However, in our previous report, insulin did not enhance the expression of CPB mRNAlevels in human hepatoma HepG2 cells.18) Therefore, the mechanism responsible for CPB gene expressiion in response to insulin might be different for 3T3-L1 adipocytes and human hepatoma HepG2 cells, although the PI3K inhibitor LY294002 suppressed the expression of the CPB2 gene in HepG2 cells. PI3K is a lipid kinase that controls multiple cellular processes through activation of Akt. Activation of the serine/threonine kinase Akt leads to multiple cellular processes, such as inhibition of apoptosis, alteration of the cell cycle and promotion of cancer invasion.32–34) It is well known that the PI3K signaling pathway is a critical transmitter of intracellular signaling in modulating platelet functions. Platelets play a critical role in atherothrombosis that can lead to ischemic stroke and myocardial infarction.35) The present results clearly showed that the PI3K inhibitor LY294002 suppressed the levels of CPB2 mRNA and TAFI antigen expression in HepG2 cells, indicating a novel therapeutic target for the prevention of thrombotic disorders through the PI3K signaling pathway. Since Akt is a downstream effector of PI3K and is thought to mediate many biological effects as described above, we also investigated the effect of an Akt inhibitor on the expression of TAFI antigen. TAFI antigen released from HepG2 cells was markedly repressed by treatment of the cells with Akt inhibitor II and the reduction of antigen levels was in parallel with that of intracellular levels of TAFI antigen in those cells (data not shown). Elevated levels of TAFI in circulating plasma are a risk factor for thrombotic and atherothrombotic diseases, such as ischemic stroke, deep vein thrombosis and coronary artery disease.12,14,36) Thus, the PI3K/Akt signaling pathway is involved in the regulation of CPB2 mRNA expression in human hepatoma cells, and may prevent thrombotic disorders through the downregulation of plasma TAFI antigen levels.
In conclusion, our results indicate that expression of CPB2 mRNA in human hepatoma HepG2 cells is downregulated by inhibition of the PI3K/Akt signaling pathway. The decrease in CPB2 mRNA levels was mediated by the enhanced instability of the message. The molecular mechanism by which the PI3K/Akt signaling pathway regulates CPB2 gene expression in liver cells will provide useful information to better understand the physiological roles of TAFI.
Acknowledgments
This work was supported in part by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors would also like to thank all of the researchers who were involved in the research.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Bouma BN, Marx PF, Mosnier LO, Meijers JC. Thrombin-activatable fibrinolysis inhibitor (TAFI, plasma procarboxypeptidase B, procarboxypeptidase R, procarboxypeptidase U). Thromb. Res., 101, 329–354 (2001).
- 2) Sakharov DV, Plow EF, Rijken DC. On the mechanism of the antifibrinolytic activity of plasma carboxypeptidase B. J. Biol. Chem., 272, 14477–14482 (1997).
- 3) Wang W, Boffa MB, Bajzar L, Walker JB, Nesheim ME. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activable fibrinolysis inhibitor. J. Biol. Chem., 273, 27176–27181 (1998).
- 4) Wu C, Dong N, da Cunha V, Martin-McNulty B, Tran K, Nagashima M, Wu Q, Morser J, Wang YX. Activated thrombin-activatable fibrinolysis inhibitor attenuates spontaneous fibrinolysis of batroxobin-induced fibrin deposition in rat lungs. Thromb. Haemost., 90, 414–421 (2003).
- 5) Muto Y, Suzuki K, Sato E, Ishii H. Carboxypeptidase B inhibitors reduce tissue factor-induced renal microthrombi in rats. Eur. J. Pharmacol., 461, 181–189 (2003).
- 6) Suzuki K, Muto Y, Fushihara K, Kanemoto K, Iida H, Sato E, Kikuchi C, Matsushima T, Kato E, Nomoto M, Yoshioka S, Ishii H. Enhancement of fibrinolysis by EF6265 [(S)-7-amino-2-[[[(R)-2-methyl-1-(3-phenylpropanoylamino)propyl]hydroxyphosphinoyl]- methyl]heptanoic acid], a specific inhibitor of plasma carboxypeptidase B. J. Pharmacol. Exp. Ther., 309, 607–615 (2004).
- 7) Campbell W, Okada N, Okada H. Carboxypeptidase R is an inactivator of complement-derived inflammatory peptides and an inhibitor of fibrinolysis. Immunol. Rev., 180, 162–167 (2001).
- 8) Tan AK, Eaton DL. Activation and characterization of procarboxypeptidase B from human plasma. Biochemistry, 34, 5811–5816 (1995).
- 9) de Bruijne EL, Gils A, Rijken DC, de Maat MP, Guimaraes AH, Poldermans D, Declerck PJ, Leebeek FW. High thrombin activatable fibrinolysis inhibitor levels are associated with an increased risk of premature peripheral arterial disease. Thromb. Res., 127, 254–258 (2011).
- 10) Eichinger S, Schonauer V, Weltermann A, Minar E, Bialonczyk C, Hirschl M, Schneider B, Quehenberger P, Kyrle PA. Thrombin-activatable fibrinolysis inhibitor and the risk for recurrent venous thromboembolism. Blood, 103, 3773–3776 (2004).
- 11) Montaner J, Ribo M, Monasterio J, Molina CA, Alvarez-Sabin J. Thrombin-activable fibrinolysis inhibitor levels in the acute phase of ischemic stroke. Stroke, 34, 1038–1040 (2003).
- 12) Santamaría A, Oliver A, Borrell M, Mateo J, Belvis R, Marti-Fabregas J, Ortin R, Tirado I, Souto JC, Fontcuberta J. Risk of ischemic stroke associated with functional thrombin-activatable fibrinolysis inhibitor plasma levels. Stroke, 34, 2387–2391 (2003).
- 13) Tregouet DA, Schnabel R, Alessi MC, Godefroy T, Declerck PJ, Nicaud V, Munzel T, Bickel C, Rupprecht HJ, Lubos E, Zeller T, Juhan-Vague I, Blankenberg S, Tiret L, Morange PE, AtheroGene Investigators. Activated thrombin activatable fibrinolysis inhibitor levels are associated with the risk of cardiovascular death in patients with coronary artery disease: the AtheroGene study. J. Thromb. Haemost., 7, 49–57 (2009).
- 14) Leebeek FW, Goor MP, Guimaraes AH, Brouwers GJ, Maat MP, Dippel DW, Rijken DC. High functional levels of thrombin-activatable fibrinolysis inhibitor are associated with an increased risk of first ischemic stroke. J. Thromb. Haemost., 3, 2211–2218 (2005).
- 15) Hori Y, Gabazza EC, Yano Y, Katsuki A, Suzuki K, Adachi Y, Sumida Y. Insulin resistance is associated with increased circulating level of thrombin-activatable fibrinolysis inhibitor in type 2 diabetic patients. J. Clin. Endocrinol. Metab., 87, 660–665 (2002).
- 16) Hori Y, Nakatani K, Morioka K, Katsuki A, Gabazza EC, Yano Y, Nobori T, Adachi Y, Sumida Y. Insulin enhanced thrombin-activable fibrinolysis inhibitor expression through PI3 kinase/Akt pathway. Int. J. Mol. Med., 15, 265–268 (2005).
- 17) Ladenvall C, Gils A, Jood K, Blomstrand C, Declerck PJ, Jern C. Thrombin activatable fibrinolysis inhibitor activation peptide shows association with all major subtypes of ischemic stroke and with TAFI gene variation. Arterioscler. Thromb. Vasc. Biol., 27, 955–962 (2007).
- 18) Ishii H, Kojima Y, Masuda Y, Takada K, Sugimoto K, Ikeda T. Expression of thrombin-activatable fibrinolysis inhibitor (TAFI) is up-regulated by increase in intracellular cyclic AMP levels in cultured HepG2 cells. Thromb. Haemost., 102, 1204–1211 (2009).
- 19) Masuda Y, Saotome D, Takada K, Sugimoto K, Sasaki T, Ishii H. Peroxisome proliferator-activated receptor-alpha agonists repress expression of thrombin-activatable fibrinolysis inhibitor by decreasing transcript stability. Thromb. Haemost., 108, 74–85 (2012).
- 20) Redlitz A, Tan AK, Eaton DL, Plow EF. Plasma carboxypeptidases as regulators of the plasminogen system. J. Clin. Invest., 96, 2534–2538 (1995).
- 21) Ishii H, Horie S, Kizaki K, Kazama M. Retinoic acid counteracts both the downregulation of thrombomodulin and the induction of tissue factor in cultured human endothelial cells exposed to tumor necrosis factor. Blood, 80, 2556–2562 (1992).
- 22) Wilson GM, Deeley RG. An episomal expression vector system for monitoring sequence-specific effects on mRNA stability in human cell lines. Plasmid, 33, 198–207 (1995).
- 23) Dani C, Piechaczyk M, Audigier Y, El Sabouty S, Cathala G, Marty L, Fort P, Blanchard JM, Jeanteur P. Characterization of the transcription products of glyceraldehyde 3-phosphate-dehydrogenase gene in HeLa cells. Eur. J. Biochem., 145, 299–304 (1984).
- 24) Broze GJ Jr, Higuchi DA. Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood, 88, 3815–3823 (1996).
- 25) Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685 (1970).
- 26) Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem., 72, 248–254 (1976).
- 27) Boffa MB, Hamill JD, Maret D, Brown D, Scott ML, Nesheim ME, Koschinsky ML. Acute phase mediators modulate thrombin-activable fibrinolysis inhibitor (TAFI) gene expression in HepG2 cells. J. Biol. Chem., 278, 9250–9257 (2003).
- 28) Garand M, Bastajian N, Nesheim ME, Boffa MB, Koschinsky ML. Molecular analysis of the human thrombin-activatable fibrinolysis inhibitor gene promoter. Br. J. Haematol., 138, 231–244 (2007).
- 29) Boffa MB, Hamill JD, Bastajian N, Dillon R, Nesheim ME, Koschinsky ML. A role for CCAAT/enhancer-binding protein in hepatic expression of thrombin-activable fibrinolysis inhibitor. J. Biol. Chem., 277, 25329–25336 (2002).
- 30) Tebo J, Der S, Frevel M, Khabar KS, Williams BR, Hamilton TA. Heterogeneity in control of mRNA stability by AU-rich elements. J. Biol. Chem., 278, 12085–12093 (2003).
- 31) Maret D, Boffa MB, Brien DF, Nesheim ME, Koschinsky ML. Role of mRNA transcript stability in modulation of expression of the gene encoding thrombin activable fibrinolysis inhibitor. J. Thromb. Haemost., 2, 1969–1979 (2004).
- 32) Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr. Cancer Drug Targets, 13, 234–244 (2013).
- 33) Tanno S, Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR. AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res., 61, 589–593 (2001).
- 34) Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W, Donowitz M, Tsichlis PN, Larue L. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res., 63, 2172–2178 (2003).
- 35) Barrett NE, Holbrook L, Jones S, Kaiser WJ, Moraes LA, Rana R, Sage T, Stanley RG, Tucker KL, Wright B, Gibbins JM. Future innovations in anti-platelet therapies. Br. J. Pharmacol., 154, 918–939 (2008).
- 36) Zorio E, Castello R, Falco C, Espana F, Osa A, Almenar L, Aznar J, Estelles A. Thrombin-activatable fibrinolysis inhibitor in young patients with myocardial infarction and its relationship with the fibrinolytic function and the protein C system. Br. J. Haematol., 122, 958–965 (2003).