RESULTS AND DISCUSSION
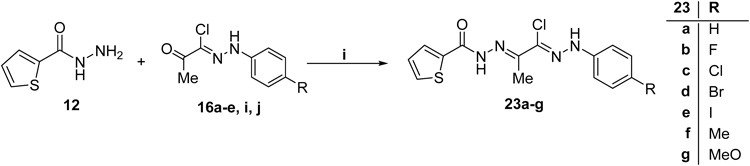
Discussion of ChemistryIn continuation of our endeavor towards the development of potent antimicrobial agents,29–33) we synthesized a series of novel twenty 5-nitrofuran-2-carbohydrazide and thiophene-2-carbohydrazide derivatives bearing different aryl and heteroaryl rings. The synthetic route was initiated with the preparation of carbohydrazides 11 and 1234) (Chart 1). Diazotization of aromatic amines 13a–j with hydrochloric acid and sodium nitrite gave diazonium salts 14a–j which subsequently coupled with 3-chloropentane-2,4-dione (15) in ethanolic sodium acetate (Japp–Klingemann reaction) to afford oxo-N-arylpropanehydrazonoyl chlorides 16a–j, respectively35) (Chart 1). Moreover, bromination of ketones 17a–e was performed using cupric bromide to yield the corresponding α-bromo ketones 18a–e,36) respectively, which then reacted with sodium benzenesulfinate (19a) or sodium toluenesulfinate (19b) to furnish the β-keto sulfones 20a–e, respectively37) (Chart 1).

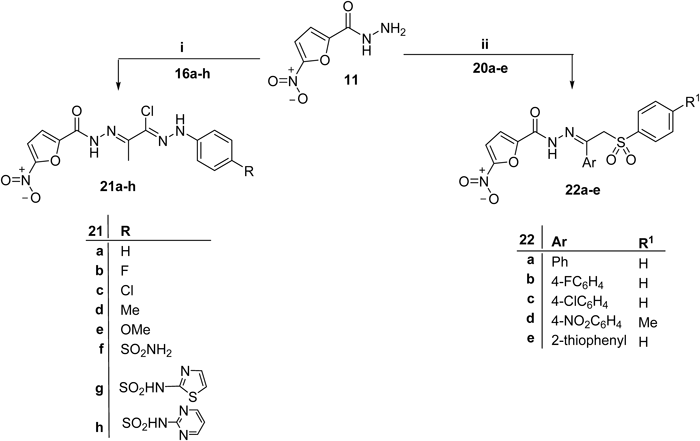
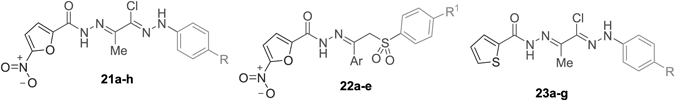
Preparation of the target compounds 21a–h was achieved via the reaction of the appropriate 2-oxo-N′-(4-substitutedphenyl)propanehydrazonoyl chloride 16a–h with 5-nitrofuran-2-carbohydrazide 11 in refluxed tetrahydrofuran (THF) (Chart 2). Next, we aimed to hybride arylsulphone moiety, as promising class of antimicrobial agents,38,39) with 5-nitrofuran scaffold through hydrazone linker. Thus, arylsulfones 20a–e were condensed with 5-nitrofuran-2-carbohydrazide (11) in ethanol, in the presence of a catalytic amount of acetic acid, to give the corresponding arylsulphones 22a–e (Chart 2).
Finally, different 2-oxo-N′-(4-substitutedphenyl)propanehydrazonoyl chlorides were refluxed with thiophene-2-carbohydrazide (12) in THF to furnish the target derivatives 23a–g (Chart 3).
IR spectra of bis-hydrazones 21a–h and 23a–g revealed the presence of stretching vibrations of the carbonyl groups in the region 1636–1696 cm−1, in addition to the absorption bands of two NH functions in the region 32465–3481 cm−1. Also their 1H-NMR spectra showed two D2O exchangeable singlet signals in the regions δ 10.05–10.96 and 10.83–11.82 ppm attributable to two NH groups, in addition to the singlet signal of methyl group in the region δ 2.35–2.40 ppm. The sulfonamide 21f and substituted sulfonamide 21g and 21h analogs displayed another D2O exchangeable signal within 7.20–12.65 ppm. Compounds 21b–d, 21f–h, 23a–c and 23e–g were confirmed by their 13C-NMR and showed a characteristic signal resonating at 152.00–188.62 ppm that confirmed the presence of carbonyl group. The analyses of the latter products confirmed the assigned structure 21a–h and 23a–g (Charts 1, 2). Recently, we reported the X-ray diffraction structure for an analogue of compounds 21, which confirmed the (1Z,2E)-configuration of these bis-hydrazones in solid state.30)
On the other hand, structures of sulfones 22a–e were characterized using their IR, 1H-NMR, and MS spectra. The IR spectra of 22a–e exhibited characteristic absorption band at 1680–1700 cm−1 due to C=O group, while that of the sulfonyl functionality was observed in the regions 1150–1158 cm−1 and 1307–1412 cm−1. Their 1H-NMR spectra exhibited the two D2O-exchangeable signal of hydrazone NH at δ 11.13–11.67 ppm. The compounds 22c–e showed characteristic 13C-NMR signal at δ 181.77–189.03 ppm attributed to the carbonyl group.
Discussion of Anti-microbial ActivityAntibacterial, antifungal and antimycobacterial activities were performed at The Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Cairo, Egypt.
Anti-fungal ActivityTarget compounds 21a–h, 22a–e, 23a–g and amphotericin, a membrane-active polyene macrolide antibiotic and an antifungal reference drug,40) were evaluated in vitro for their antifungal activity by inhibition zone technique and minimum inhibitory concentration (MIC). Data in Tables 1 and 2 revealed that all the compounds showed no activity against Candida albicans, while compounds 21b, f, g and 22b showed a remarkable activity against Aspergillus fumigatus.
Table 1. Antimicrobial Activity of the Synthesized Compounds against the Pathological Organisms Expressed as Inhibition Diameter Zones in Millimeters (mm) Based on Well Diffusion Assay
 |
| Compd | Ar | R or R1 | Fungi | Gram-positive bacteria | Gram-negative bacteria |
|---|
| Af | Ca | Sa | Sp | Bs | Pa | St | Kp | Ec |
|---|
| 21a | | H | 18.3±0.58 | NA | 20.4±0.58 | 20.9±0.44 | 22.3±0.44 | NA | 20.3±0.58 | 21.6±0.37 | 19.6±0.58 |
| 21b | | F | 20.6±0.44 | NA | 20.4±0.35 | 21.2±0.44 | 22.3±0.25 | 19.2±0.25 | 20.6±0.37 | 22.4±0.63 | 20.3±0.63 |
| 21c | | Cl | 16.4±0.25 | NA | NA | NA | NA | NA | NA | NA | NA |
| 21d | | Me | 15.2±0.58 | NA | 16.2±1.2 | 17.1±0.58 | 17.9±2.1 | NA | 16.2±0.58 | 16.4±0.63 | 18.1±0.58 |
| 21e | | OMe | 14.6±0.63 | NA | 16.7±0.72 | 18.2±0.44 | 19.3±0.36 | NA | 17.4±0.58 | 19.3±0.63 | 16.2±0.58 |
| 21f | | SO2NH2 | 22.3±0.58 | NA | 24.1±0.44 | 24.9±0.63 | 25.2±0.58 | NA | 21.4±0.37 | 22.6±0.63 | 21.3±0.63 |
| 21g | |  | 20.3±0.44 | NA | 21.6±0.63 | 23.8±1.2 | 23.9±0.44 | NA | 20.4±0.25 | 21.3±1.2 | 18.8±0.58 |
| 21h | | 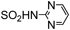 | 18.1±0.72 | NA | 20.2±0.72 | 18.6±0.58 | 21.8±0.58 | NA | 20.9±0.58 | 22.0±1.2 | 20.2±0.63 |
| 22a | Ph | H | 16.3±0.25 | NA | 18.2±1.2 | 20.3±0.37 | 21.4±0.63 | NA | 20.3±0.48 | 21.2±0.63 | 18.5±0.58 |
| 22b | 4-FC6H4 | H | 18.6±1.2 | NA | 20.4±1.2 | 22.3±0.63 | 22.4±0.58 | NA | 19.3±0.63 | 20.4±0.63 | 19.1±1.2 |
| 22c | 4-ClC6H4 | H | 17.2±1.2 | NA | 19.3±0.63 | 18.1±0.58 | 21.2±0.72 | NA | 20.1±0.58 | 21.0±0.63 | 19.2±0.58 |
| 22d | 4-NO2C6H4 | CH3 | 13.2±1.2 | NA | 15.1±0.44 | 16.2±0.44 | 16.9±0.72 | NA | 15.1±0.58 | 15.9±0.72 | 13.1±1.2 |
| 22e | 2-Thiophenyl | H | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23a | | H | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23b | | F | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23c | | Cl | 17.2±0.72 | NA | NA | NA | NA | NA | NA | NA | NA |
| 23d | | Br | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23e | | I | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23f | | Me | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23g | | OMe | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| AB | | | 20.4±0.24 | 21.6±0.22 | | | | | | | |
| CF | | | | | 20.2±0.34 | 20.3±0.58 | 20.4±0.14 | 19.1±0.15 | 19.8± 0.63 | 19.7±0.12 | 20.3± 0.44 |
NA: No activity. The screening organisms, Mould: Aspergillus fumigatus (RCMB 02568, An), An Yeasts: Candida albicans (RCMB 05036, Ca), Gram-positive bacteria: Staphylococcus aureus (RCMB 010028, Sa), Streptococus pneumoniae (RCMB 010010, Sp), and Bacillus subtilis (RCMB 010069, Bs). Gram-negative bacteria: Pseudomonas aeruginosa (RCMB 010043, Pa), Salmonella typhimurium (RCMB 010315, St), Klebsiella penumoniae (RCMB 0010093, Kp) and Escherichia coli (RCMB 010052, Ec), AB: Amphotericin B, CF: Ciprofloxacin.
Table 2. Antimicrobial Activity as MICs (µg/mL) of Tested Standards and Synthesized Compounds against Tested Microorganisms
| Compd | Fungi | Gram-positive bacteria | Gram-negative bacteria |
|---|
| Af | Ca | Sa | Sp | Bs | Pa | St | Kp | Ec |
|---|
| 21a | 7.81 | NA | 1.95 | 0.98 | 0.49 | NA | 1.95 | 0.98 | 3.90 |
| 21b | 0.98 | NA | 1.95 | 0.98 | 0.49 | 3.90 | 0.98 | 0.49 | 1.95 |
| 21c | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 21d | 62.5 | NA | 31.25 | 15.63 | 7.81 | NA | 31.25 | 31.25 | 7.81 |
| 21e | 62.5 | NA | 15.63 | 7.81 | 3.90 | NA | 7.81 | 3.90 | 31.25 |
| 21f | 0.49 | NA | 0.12 | 0.06 | 0.06 | NA | 0.98 | 0.24 | 0.98 |
| 21g | 1.95 | NA | 0.49 | 0.12 | 0.12 | NA | 1.95 | 0.98 | 3.90 |
| 21h | 7.81 | NA | 1.95 | 3.90 | 0.49 | NA | 0.98 | 0.49 | 1.95 |
| 22a | 31.25 | NA | 7.81 | 1.95 | 0.98 | NA | 1.95 | 0.98 | 3.90 |
| 22b | 3.90 | NA | 1.95 | 0.49 | 0.49 | NA | 1.95 | 1.95 | 3.90 |
| 22c | 15.63 | NA | 3.90 | 7.81 | 0.98 | NA | 1.95 | 0.98 | 3.90 |
| 22d | 125 | NA | 62.50 | 31.25 | 15.63 | NA | 62.50 | 31.25 | 125 |
| 22e | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23a | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23b | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23c | 15.63 | NA | NA | NA | NA | NA | NA | NA | NA |
| 23d | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23e | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23f | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 23g | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| AB | 1.95 | 0.49 | | | | | | | |
| CF | | | 1.95 | 1.95 | 1.95 | 3.90 | 3.90 | 3.90 | 1.95 |
NA: No Activity. Results in bold indicate more or equal activity relative to standard drugs. The screening organisms, Mould: Aspergillus fumigatus (RCMB 02568, An), An Yeasts: Candida albicans (RCMB 05036, Ca), Gram-positive bacteria: Staphylococcus aureus (RCMB 010028, Sa), Streptococus pneumoniae (RCMB 010010, Sp), and Bacillus subtilis (RCMB 010069, Bs). Gram-negative bacteria: Pseudomonas aeruginosa (RCMB 010043, Pa), Salmonella typhimurium (RCMB 010315, St), Klebsiella penumoniae (RCMB 0010093, Kp) and Escherichia coli (RCMB 010052, Ec), AB: Amphotericin B, CF: Ciprofloxacin.
Aspergillus fumigatus is largely responsible for increasing the mortality rate in immunocompromised patients due to invasive aspergillosis (IA).41) The main reason for this unacceptable mortality rate is the limited number of antifungal agents.42) Whilst, the thiophene containing counterparts 22e and 23a–g displayed no activity, the 5-nitrofuran derivatives 21a–h and 22a–d showed moderate to excellent activity toward the Aspergillus fumigatus with MIC values ranging from 0.49 to 125 µg/mL. In particular, the sulfonamide derivative 21f was found to be the most potent counterpart against Aspergillus fumigatus organism (MIC=0.49 µg/mL) as it was four times more active than the reference drug Amphotericin B (MIC=1.95 µg/mL).
Antimycobacterial and in Vitro Cytotoxic ActivitiesAntitubercular activity of the newly synthesized derivatives 21a–h, 22a–e and 23a–g against M. tuberculosis (RCMB 010126) was evaluated at The Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Cairo, Egypt. The mean values of the inhibition zone diameter and the minimum inhibitory concentrations were listed in Table 3. Isoniazide and pyrazinamide were included in the experiments as reference drugs (MIC=0.40 and 3.21 µg/mL, respectively).
Table 3. Anti-tubercular (MIC), Cytotoxicity (IC
50) and Selectivity Index (SI) of the Synthesized Compounds
| Compd | I.Z | MICa) | IC50b) | SIc) |
|---|
| 21a | 17.6±0.37 | 7.81 | >50 | >6.4 |
| 21b | 16.3±0.58 | 31.25 | n.d. | n.d. |
| 21c | NA | NA | n.d. | n.d. |
| 21d | 14.3±1.2 | 125 | n.d. | n.d. |
| 21e | 15.2±0.58 | 62.50 | n.d. | n.d. |
| 21f | 19.6±0.58 | 3.90 | >50 | >12.8 |
| 21g | 18.1±0.58 | 7.81 | >50 | >6.4 |
| 21h | 17.1±0.58 | 15.63 | 33.70±0.03 | 2.16 |
| 22a | 17.2±1.4 | 15.63 | >50 | >3.2 |
| 22b | 17.6±0.44 | 7.81 | >50 | >6.4 |
| 22c | 16.6±0.73 | 15.63 | >50 | >3.2 |
| 22d | 14.3±0.63 | 125 | n.d. | n.d. |
| 22e | NA | NA | n.d. | n.d. |
| 23a | NA | NA | n.d. | n.d. |
| 23b | NA | NA | n.d. | n.d. |
| 23c | NA | NA | n.d. | n.d. |
| 23d | NA | NA | n.d. | n.d. |
| 23e | NA | NA | n.d. | n.d. |
| 23f | NA | NA | n.d. | n.d. |
| 23g | NA | NA | n.d. | n.d. |
| Isoniazide | | 0.40 | | |
| Pyrazinamide | | 3.21 | | |
| Doxorubicin | | | 0.31±0.09 | |
a) Minimum inhibitory concentration of M. tuberculosis (µg/mL). b) Measurement of cytotoxicity in breast MDA-MB-231 cells: 50% inhibitory concentrations (µg/mL). c) Selectivity index (in vitro): IC50 in breast MDA-MB-231/MIC against M. tuberculosis. NA: No Activity, n.d.: not determined.
Analysis of the data in Table 3 illustrated that compounds of the first series 21a–h and second series 22a–e displayed good to fair antimycobacterial activity, while the third series derivatives 23a–g lacked the activity, in a similar fashion to the antifungal and antibacterial activities. Compound 21f, which possessed superior antifungal and antibacterial activities, exploited the most potent antitubercular activity (I.Z=19.6±0.58 mm and MIC=3.90 µg/mL), confirming the significance of the incorporation of the sulfonamido functionality. In addition, compounds 21a, b, g, h and 22a–c showed moderate antitubercular activity with MIC values ranging from 7.81 to 31.25 µg/mL.
In vitro cytotoxicity of the most active antimicrobial and antitubercular compounds 21a, f–g and 22a–c was evaluated against human breast MDA-MB-231 cells using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Doxorubicin was used as the positive control drug. The IC50 values obtained for these compounds are listed in Table 3. None of the tested compounds displayed any significant cytotoxicity, thereby providing a high therapeutic index. Selectivity index (SI) is used to estimate the therapeutic effect of a drug and to identify drug candidates for further studies. SI of each compound was determined as the ratio of IC50 to MIC (Table 3). It was reported that, new drugs candidates must have SI equal or higher than 1.43) In this study, 21f could be considered as a promising new anti-tubercular drug candidate with SI >12.8.
Antibacterial ActivityInitially, all the synthesized compounds (21a–h, 22a–e and 23a–g) were evaluated in vitro for their antibacterial activity, by inhibition zone technique, using three Gram-positive bacteria that are Staphylococcus aureus (RCMB 010028), Streptococus pneumoniae (RCMB 010010), and Bacillus subtilis (RCMB 010069) and four Gram-negative bacteria that are Pseudomonas aeruginosa (RCMB 010043), Salmonella typhimurium (RCMB 010315), Klebsiella penumoniae (RCMB 0010093) and Escherichia coli (RCMB 010052), as presented in Table 1. Subsequently, MIC of all the synthesized compounds was evaluated in vitro using the twofold serial dilution technique. The lowest concentration showing no growth was taken as the MIC. The results of minimum inhibitory concentration were reported in Table 2. Ciprofloxacin, a broad-spectrum widely used antibacterial and the most potent marketed fluoroquinolone against Gram-negative bacteria,12) was used as reference drug in this assay.
As shown in Tables 1 and 2, the tested compounds displayed different levels of antibacterial activity and possessed a distinctive pattern of selectivity against the tested strains. With the exception of compounds 21c and 22e, all compounds of the first and second series showed a remarkable broad-spectrum antibacterial activity against almost all tested strains used in the assay. On the other hand, the remaining compounds 23a–g had no significant activity against any of the tested strains at concentration up to 125 µg/mL. All the tested Gram-positive and Gram-negative strains were susceptible to the all active compounds influence, except P. aeruginosa affected only with compound 21b. Investigation of the activity against B. subtilis indicated that it was the most sensitive strain to the influence of the active members.
Observing the results, we could deduce valuable data about the structure activity correlation of the tested compounds. Firstly, we explored the impact of substitution of the 4-position of the pendant phenyl group in the first series compounds 21a–h. Incorporation of unsubstituted phenyl group led to compound 21a with broad and excellent activity against S. aureus, S. pneumonia, B. subtilis, S. typhimurium, K. pneumonia and E. coli (MIC=0.49–3.9 µg/mL). Since fluorine has a size and electronic properties similar to those of hydrogen, it is introduced as an isosteric to the hydrogen atom. Compound 21b bearing fluorine substituent at the 4-position, showed an increase in the activity against the tested Gram-negative bacteria, especially against P. aeruginosa (MIC=3.9 µg/mL). Also, it retained the activity of the Gram-positive one suggesting that the substitution in the 4-position may be tolerated also suggesting that the halogens incorporation may be advantageous. Conversely, substitution with more bulky chlorine atom, compound 21c, abolished the activity. Furthermore, grafting methyl group, compound 21d, and methoxy group, compound 21e, resulted in drop of the activity (MIC=3.9–31.25 µg/mL).
Concerning the effect of the substitution in the first series member’s 21a–e, the activities were decreased in the order of 4-F>unsubstituted>4-OMe>4-Me>>>4-Cl, hinting that grafting a small electron-withdrawing group like fluorine is more beneficial than an electron-donating group like methyl or methoxy for the antibacterial activity.
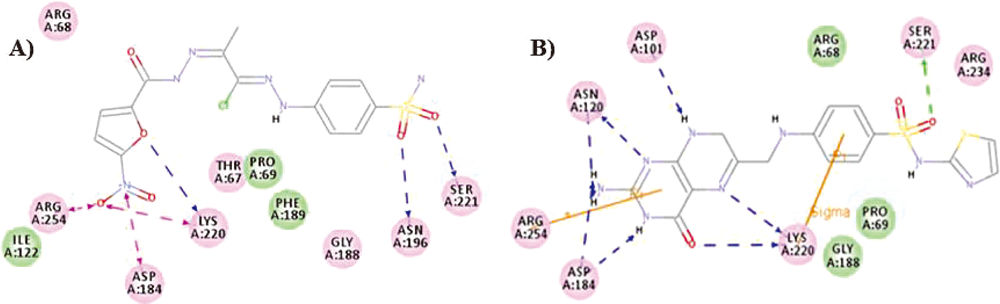
Taking into consideration the importance of sulfonamido substituent as antimicrobial pharmacophore,44) 4-sulfonamido functionalities were incorporated in 21f–g counterparts. Unsubstituted sulfonamide derivative 21f emerged as the most potent analog with outstanding antimicrobial activities (MIC=0.06–0.98 µg/mL), confirming the importance of sulfonamido substitution. Notably, 21f analog displayed more susceptibility towards Gram-positive bacteria (MIC=0.06–0.12 µg/mL) more than Gram-negative one (MIC=0.24–0.98 µg/mL), relative to the standard drug. Substitution with more bulky sulfonamide derivatives N-(thiazol-2-yl)sulfonamido and N-(pyrimidin-2-yl)sulfonamido produced compounds 21g and h with potent and broad-spectrum activity (MIC=0.12–3.9 µg/mL), respectively. Thence, the order of activities of the sulfonamides members in the first series, were decreased in the order of sulfanilamide>sulfathiazole>sulfadiazine for the Gram-positive strains, and in the order of sulfanilamide>sulfadiazine>sulfathiazole for the Gram-positive bacteria. Moreover, a molecular docking study was used to explain the obtained excellent biological data of 21f.
Considering the activities of 5-nitrofuran analogs containing sulfone group 22a–e, 4-F substituted analog 22b exhibited superior activity against Gram-positive bacteria (S. aureus, S. pneumonia and B. subtilis) with MIC values of 1.95, 0.49 and 0.49 µg/mL, respectively, confirming the particularity of the incorporation of small electron-withdrawing group. In addition, the unsubstituted 22a and the 4-Cl substituted 22c derivatives elicited good antibacterial activity against the tested strains (MIC=0.98–7.81 µg/mL). On the other hand, compound 22d possessed modest activity against the tested bacteria (MIC=15.63–125 µg/mL). Also, it was found that the replacement of the phenyl group with 2-thiophenyl one, as compound 22e, led to complete loss of activity.
Interestingly, bioisosteric replacement of the 5-nitrofuran moiety with thiophene one in the third series led up to diminished activity, confirming the role of 5-nitrofuran group as a crucial element for the antibacterial activity.
MATERIALS AND METHODS
General ChemistryMelting points (mp) were measured with a Stuart melting point apparatus and were uncorrected. The NMR spectra were recorded by Varian Gemini-300BB 300 MHz FT-NMR spectrometers (Varian Inc., Palo Alto, CA, U.S.A.). 1H and 13C spectra were run at 500 and 125 MHz, respectively, in deuterated dimethyl sulfoxide (DMSO-d6). Chemical shifts (δH) are reported relative to tetramethylsilane, as internal standard. Electron impact mass spectra were measured on a Varian MAT 311-A (70 eV). Reaction courses and product mixtures were routinely monitored by thin layer chromatography (TLC) on silica gel precoated F254 Merck plates. Unless otherwise noted, all solvents and reagents were commercially available and used without further purification.
Synthesis of Hydrazones 21a–hTo a stirred solution of the 5-nitrofuran-2-carbohydrazide 11 (5 mmol), 2-oxo-N′-(4-substitutedphenyl)propanehydrazonoyl chloride 16a–h (5 mmol) was added in THF (30 mL). The reaction mixture was heated under reflux for 10 h. The solid product obtained upon cooling was filtered off and recrystallized from dioxan to afford the corresponding hydrazones 21a–h with 65–80% yield.
(1Z,2E)-2-(2-(5-Nitrofuran-2-carbonyl)hydrazono)-N′-phenylpropanehydrazonoyl Chloride (21a)Orange powder (yield 75%), mp 230°C; IR (KBr, ν cm−1): 3414, 3315 (2NH), 1668 (C=O) and 1599 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.38 (s, 3H, CH3), 6.94–7.98 (m, 7H, ArH), 10.27 (s, D2O exch., 1H, =NNH–), 11.10 and 11.55 (s, D2O exch., 1H, –CONH–); MS m/z [%]: 351 [(M+2)+, 9.3], 349 [M+, 26.9], 92 [100].
(1Z,2E)-N′-(4-Fluorophenyl)-2-(2-(5-nitrofuran-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (21b)Orange powder (yield 75%), mp 232°C; IR (KBr, ν cm−1): 3420, 3317 (2NH), 1652 (C=O) and 1589 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 7.13 (t, J=7.5 Hz, 2H, Ar-H), 7.36–7.97 (m, 4H, ArH), 10.29 (s, D2O exch., 1H, =NNH–), 11.10 and 11.54 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 19.02, 113.48, 115.66 (3JF-C=7.5 Hz), 116.11(2JF-C=22.5 Hz), 122.15, 123.50, 127.00, 140.46 (2C), 152.53, 156.83 (1JF-C=236.3 Hz), 157.41; MS m/z [%]: 369 [(M+2)+, 7.0], 367 [M+, 18.9], 110 [100].
(1Z,2E)-N′-(4-Chlorophenyl)-2-(2-(5-nitrofuran-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (21c)Red powder (yield 75%), mp 231°C; IR (KBr, ν cm−1): 3414, 3319 (2NH), 1669 (C=O) and 1595 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 7.35–7.96 (m, 6H, ArH), 10.39 (s, D2O exch., 1H, =NNH–), 11.15 and 11.56 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 13.88, 113.47, 115.94, 122.13, 125.32, 129.46, 142.86, 146.91, 152.56, 147.33, 148.81, 154.89; MS m/z [%]: 387 [(M+4)+, 1.4 ], 385 [(M+2)+, 8.3], 383 [M+, 12.6], 126 [100].
(1Z,2E)-2-(2-(5-Nitrofuran-2-carbonyl)hydrazono)-N′-p-tolylpropanehydrazonoyl Chloride (21d)Red powder (yield 75%), mp 239°C; IR (KBr, ν cm−1): 3420, 3315 (2NH), 1668 (C=O) and 1576 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.25 (s, 3H, CH3), 2.35 (s, 3H, CH3), 7.10 (d, J=6 Hz, 2H, Ar-H), 7.24 (d, J=6 Hz, 2H, Ar-H), 7.68–7.97 (m, 2H, H3 and H4 of furan), 10.17 (s, D2O exch., 1H, =NNH–), 11.09 and 11.52 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 13.82, 20.76, 113.47, 114.39, 115.32, 122.43, 130.02, 141.57, 146.47, 147.11, 147.40, 152.49, 157.40; MS m/z [%]: 365 [(M+2)+, 9.8], 363 [M+, 28.8], 106 [100].
(1Z,2E)-N′-(4-Methoxyphenyl)-2-(2-(5-nitrofuran-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (21e)Brown powder (yield 75%), mp 224°C; IR (KBr, ν cm−1): 3414, 3318 (2NH), 1668 (C=O) and 1576 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.35 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 6.90 (d, 2H, Ar-H, J=7.5 Hz), 7.29 (d, 2H, Ar-H, J=7.5 Hz), 7.70–7.98 (m, 2H, H3 and H4 of furan), 10.13 (s, D2O exch., 1H, =NNH–), 11.10 and 11.51 (s, D2O exch., 1H, –CONH–); MS m/z [%]: 381 [(M+2)+, 3.5], 379 [M+, 10.2], 122 [100].
(1Z,2E)-2-(2-(5-Nitrofuran-2-carbonyl)hydrazono)-N′-(4-sulfamoylphenyl)propanehydrazonoyl Chloride (21f)Red powder (yield 75%), mp 260°C; IR (KBr, ν cm−1): 3390–3264 (2NH,NH2), 1670 (C=O) and 1596 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.37 (s, 3H, CH3), 7.20 (s, D2O exch., 2H, SO2NH2), 7.46 (d, J=7.5 Hz, 2H, Ar H), 7.75–7.95 (m, 4H, ArH), 10.59 (s, D2O exch., 1H, =NNH–), 11.14 and 11.57 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 19.00, 113.98, 115.01, 122.20, 125.29, 127.74, 136.75, 138.19, 145.66, 152.51, 157.46, 188.56; MS m/z [%]: 456 [(M+2)+, 4.6], 454 [M+, 11.8], 105 [100]; MS m/z [%]: 430 [(M+2)+, 1.2], 428 [M+, 3.0], 64 [100].
(1Z,2E)-2-(2-(5-Nitrofuran-2-carbonyl)hydrazono)-N′-(4-(N-thiazol-2-ylsulfamoyl)phenyl)propanehydrazonoyl Chloride (21g)Orange powder (yield 75%), mp 233°C; IR (KBr, ν cm−1): 3419–3253 (3NH), 1682 (C=O) and 1597 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.38 (s, 3H, CH3), 6.82 (d, J=3 Hz, 1H, H5 of thiazole), 7.25 (d, J=3 Hz, 1H, H4 of thiazole), 7.45 (d, J=7.5 Hz, 2H, Ar-H), 7.54-7.96 (m, 4H, ArH), 10.63 (s, D2O exch., 1H, =NNH–), 10.96 and 11.61 (s, D2O exch., 1H, –CONH–), 12.65 (s, D2O exch., 1H, SO2NH-); 13C-NMR (DSMO-d6) δ ppm: 25.90, 108.41, 113.50, 114.01, 115.04, 124.86, 125.14, 127.99, 134.77, 136.24, 145.90, 146.74, 153.50, 156.00, 188.57; MS m/z [%]: 511 [M+, 0.1], 111 [100].
(1Z,2E)-2-(2-(5-Nitrofuran-2-carbonyl)hydrazono)-N'-(4-(N-pyrimidin-2-ylsulfamoyl)phenyl)propanehydrazonoyl Chloride (21h)Orange powder (yield 75%), mp 236°C; IR (KBr, ν cm−1): 3481–3246 (3NH), 1696 (C=O) and 1597 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.38 (s, 3H, CH3), 7.05 (m, 1H, Ar-H), 7.47 (d, J=8.25 Hz, 1H, H4 of furan), 7.57 (d, J=8.5 Hz, 2H, Ar-H), 7.81 (s, D2O exch., 1H, SO2NH-), 7.91 (d, J=8.25 Hz, 1H, H5 of furan), 7.95 (d, J=8.5 Hz, 2H, Ar-H), 8.51 (d, J=7.0 Hz, 2H, Ar-H), 10.69 (s, D2O exch., 1H, =NNH–), 11.00 and 11.62 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 25.92, 113.80, 114.83, 116.24, 125.53, 129.00, 129.96, 132.25, 133.82, 146.59, 147.42, 157.42, 157.48, 158.82, 188.62; MS m/z [%]: 506 [M+, 2.4], 64 [100].
Synthesis of Target Compounds 22a–eThe sulfone derivatives 20a–e (1 mmol) was added to a suspension of the 5-nitrofuran-2-carbohydrazide 11 (1 mmol) in absolute ethanol (10 mL), and glacial acetic acid (0.5 mL) was added to the mixture. The reaction mixture refluxed for 0.5 h. The precipitate formed was collected by filtration while hot, washed with hot ethanol, dried and crystallized from EtOH/DMF to afford compounds 22a–e.
(E)-5-Nitro-N′-(1-phenyl-2-(phenylsulfonyl)ethylidene)furan-2-carbohydrazide (22a)Yellow powder (yield 75%), mp 233°C; IR (KBr, ν cm−1): 3420, 3278 (2NH), 1700 (C=O), 1591 (C=N) and 1354, 1150 (SO2); 1H-NMR (DMSO-d6) δ ppm: 5.36 and 5.44 (s, 2H, CH2), 7.40–7.91 (m, 12H, ArH), 11.14 and 11.66 (s, D2O exch., 1H, –CONH–); MS m/z [%]: 414 [(M+1)+, 1.1], 413 [M+, 3.2], 272 [100].
(E)-N′-(1-(4-Fluorophenyl)-2-(Phenylsulfonyl)ethylidene)-5-nitrofuran-2-carbohydrazide (22b)Yellow powder (yield 75%), mp 255°C; IR (KBr, ν cm−1): 3419, 3284 (2NH), 1700 (C=O), 1590 (C=N) and 1307, 1154 (SO2); 1H-NMR (DMSO-d6) δ ppm: 5.36 and 5.46 (s, 2H, CH2), 7.22–7.89 (m, 11H, ArH), 11.13 and 11.67 (s, D2O exch., 1H, –CONH–); MS m/z [%]: 432 [(M+1)+, 1.6], 431 [M+, 5.0], 290 [100].
(E)-N′-(1-(4-Chlorophenyl)-2-(phenylsulfonyl)ethylidene)-5-nitrofuran-2-carbohydrazide (22c)Yellow powder (yield 75%), mp 223°C; IR (KBr, ν cm−1): 3414, 3289 (2NH), 1697 (C=O), 1592 (C=N) and 1353, 1153 (SO2); 1H-NMR (DMSO-d6) δ ppm: 5.36 and 5.45 (s, 2H, CH2), 7.46–7.97 (m, 11H, ArH), 11.14 and 11.67 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 62.70, 114.50, 119.00, 128.48, 128.66, 128.80, 129.34, 129.55, 129.70, 131.42, 134.51, 134.88, 139.84, 152.49, 188.62; MS m/z [%]: 449 [(M+2)+, 2.6], 447 [M+, 6.0], 139 [100].
(E)-5-Nitro-N′-(1-(4-nitrophenyl)-2-tosylethylidene)furan-2-carbohydrazide (22d)Yellow powder (yield 75%), mp 203°C; IR (KBr, ν cm−1): 3420, 3288 (2NH), 1680 (C=O), 1595 (C=N) and 1347, 1152 (SO2); 1H-NMR (DMSO-d6) δ ppm: 2.29 (s, 3H, CH3), 5.41 (s, 2H, CH2), 7.34 (d, J=8.0 Hz, 1H, H4 of furan), 7.43 (d, J=7.0 Hz, 2H, Ar-H), 7.76 –8.07 (m, 3H, ArH), 8.18 (d, J=7.75 Hz, 2H, Ar-H), 8.32 (d, J=7.75 Hz, 2H, Ar-H), 11.13 and 11.61 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 21.40, 63.30, 123.80, 124.19, 128.57, 128.75, 129.03, 130.18, 130.93, 136.10, 136.80, 140.62, 145.30, 150.80, 154.00, 189.03; MS m/z [%]: 472 [M+, 0.6], 140 [100].
(Z)-5-Nitro-N′-(2-(phenylsulfonyl)-1-(thiophen-2-yl)ethylidene)furan-2-carbohydrazide (22e)Yellow powder (yield 75%), mp 140–150°C; IR (KBr, ν cm−1): 3414, 3292 (2NH), 1700 (C=O), 1559 (C=N) and 1412, 1158 (SO2); 1H-NMR (DMSO-d6) δ ppm: 5.25 (s, 2H, CH2), 7.25–8.10 (m, 10H, ArH), 11.13 and 11.61 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 63.04, 128.53, 128.73, 129.48, 129.70, 134.54, 137.11, 137.76, 139.73, 143.58, 181.77; MS m/z [%]: 420 [(M+1)+, 2.7], 419 [M+, 10.1], 109 [100].
Synthesis of Hydrazones 23a–gTo a stirred solution of the appropriate 2-oxo-N′-(4-substitutedphenyl)propanehydrazonoyl chloride 16a–e, i, j (5 mmol) in THF (20 mL), thiophene-2-carbohydrazide 12 (5 mmol) was added. The reaction mixture was heated under reflux for 10 h. The solid product obtained upon cooling was filtered off and recrystallized from dioxan to afford the corresponding hydrazones 23a–g with 65–80% yield.
(1Z,2E)-N′-Phenyl-2-(2-(thiophene-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (23a)Yellow powder (yield 75%), mp 220°C; IR (KBr, ν cm−1): 3414, 3325 (2NH), 1636 (C=O) and 1598 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.38 (s, 3H, CH3), 6.93–7.95 (m, 8H, ArH), 10.18 (s, D2O exch., 1H, =NNH–), 11.00 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 13.95, 114.31 (2C), 121.65, 123.81, 127.63, 129.62, 134.56 (2C), 135.32, 143.96, 152.00; MS m/z [%]: 322 [(M+2)+, 3.7], 320 [M+, 9.5], 284 [100].
(1Z,2E)-N′-(4-Fluorophenyl)-2-(2-(thiophene-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (23b)Yellow powder (yield 75%), mp 225°C; IR (KBr, ν cm−1): 3420, 3312 (2NH), 1653 (C=O) and 1592 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.37 (s, 3H, CH3), 7.13–8.20 (m, 7H, ArH), 10.22 (s, D2O exch., 1H, =NNH–), 11.00 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 13.98, 115.56 (3JF-C=7.5 Hz), 116.09 (2JF-C=22.5 Hz), 123.81, 126.50, 127.57 (2C), 133.00, 135.32 (2C), 140.61, 156.73 (1JF-C=235 Hz); MS m/z [%]: 340 [(M+2)+, 1.9], 338 [M+, 4.9], 111 [100].
(1Z,2E)-N′-(4-Chlorophenyl)-2-(2-(Thiophene-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (23c)Yellow powder (yield 75%), mp 230°C; IR (KBr, ν cm−1): 3419, 3320 (2NH), 1655 (C=O) and 1586 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.38 (s, 3H, CH3), 7.24–8.20 (m, 7H, ArH), 10.29 (s, D2O exch., 1H, =NNH–), 11.02 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 14.00, 115.86 (2C), 116.50 (2C), 124.63, 125.13, 127.60, 129.44, 135.26 (3C), 143.00, 154.00; MS m/z [%]: 356 [(M+2)+, 2.3], 354 [M+, 3.4], 111 [100].
(1Z,2E)-N′-(4-Bromophenyl)-2-(2-(thiophene-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (23d)Yellow powder (yield 75%), mp 224°C; IR (KBr, ν cm−1): 3414, 3316 (2NH), 1663 (C=O) and 1582 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.37 (s, 3H, CH3), 7.23 (d, J=3.0 Hz, 1H, H4 of thiophene), 7.31 (d, J=7.25 Hz, 2H, Ar-H), 7.45 (d, J=7.25 Hz, 2H, Ar-H), 7.95–8.20 (m, 2H, H3 and H5 of thiophene), 10.33 (s, D2O exch., 1H, =NNH–), 11.02 (s, D2O exch., 1H, –CONH–); MS m/z [%]: 402 [(M+2)+, 0.7], 400 [M+, 2.7], 111 [100].
(1Z,2E)-N′-(4-Iodophenyl)-2-(2-(thiophene-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (23e)Orange powder (yield 75%), mp 178°C; IR (KBr, ν cm−1): 3420, 3320 (2NH), 1652 (C=O) and 1586 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.40 (s, 3H, CH3), 7.10–7.91 (m, 7H, ArH), 10.08 and 10.25 (s, D2O exch., 1H, =NNH–), 10.83 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 14.31, 114.29, 114.47, 123.21, 128.73, 129.15, 130.02, 130.36, 132.02, 134.29, 141.57, 154.48; MS m/z [%]: 446 [M+, 0.3], 111 [105].
(1Z,2E)-2-(2-(Thiophene-2-carbonyl)hydrazono)-N′-p-tolylpropanehydrazonoyl Chloride (23f)Yellow powder (yield 75%), mp 221°C; IR (KBr, ν cm−1): 3420, 3316 (2NH), 1652 (C=O) and 1558 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.25 (s, 3H, CH3), 2.37 (s, 3H, CH3), 7.10 (d, J=7.50 Hz, 2H, Ar-H), 7.15–8.21 (m, 5H, ArH), 10.09 (s, D2O exch., 1H, =NNH–), 10.98 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 13.97, 20.77, 114.32 (2C), 115.32, 123.18, 127.58, 130.02 (2C), 130.39, 135.30, 141.71 (2C); MS m/z [%]: 336 [(M+2)+, 4.1], 334 [M+, 10.6], 111 [100].
(1Z,2E)-N′-(4-Methoxyphenyl)-2-(2-(thiophene-2-carbonyl)hydrazono)propanehydrazonoyl Chloride (23g)Yellow powder (yield 75%), mp 229°C; IR (KBr, ν cm−1): 3421, 3319 (2NH), 1654 (C=O) and 1564 (C=N); 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 6.90 (d, J=8.5 Hz, 2H, Ar-H), 7.23–7.30 (m, 3H, H4 thiophene +2 Ar-H), 7.95–8.25 (m, 2H, H3 and H5 thiophene), 10.05 (s, D2O exch., 1H, =NNH–), 10.95 (s, D2O exch., 1H, –CONH–); 13C-NMR (DSMO-d6) δ ppm: 13.94, 55.73, 114.98 (2C), 115.47 (2C), 122.59, 127.55, 135.28, 137.77 (2C), 154.66.
Antimicrobial ActivityAll strains were provided from culture collection of the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Cairo, Egypt. Antibacterial and antifungal activities were expressed as the diameter of inhibition zones; agar well diffusion method was used. Holes (1 cm diameter) were digger in the agar using sterile cork borer in sterile malt agar plates for fungi and sterile nutrient agar plates for bacteria, which had previously been uniformly seeded with tested microorganisms. The holes were filled by fungal filtrates (100 µL). Plates were left in a cooled incubator at 4°C for one hour for diffusion and then incubated at 37°C for tested bacteria and 28°C for tested fungi. Inhibition zones developed due to active antimicrobial metabolites were measured after 24 h of incubation for bacteria and 48 h of incubation for fungi. Amphotericin B and ciprofloxacin were used as antifungal and antibacterial positive control; respectively. The experiment was performed in triplicate and the average zone of inhibition was calculated.
Minimum Inhibitory ConcentrationMIC was performed by a serial dilution technique described by Irobi et al.,53) starting with 100 mmol concentration of all compounds dissolved in 1 mL DMSO and then reduced by successive twofold dilutions of stock solution using a calibrated micropipette. Amphotericin B and ciprofloxacin were used as the reference compounds for fungi and bacteria; respectively. The final solutions concentrations were 125, 62.50, 31.25, 15.63, 7.81, 3.90, 1.95, 0.98, 0.49, 0.24, 0.12 and 0.06 µmol/mL. The microtiter plates were incubated at 37°C for tested bacteria and 28°C for tested fungi and were readied using microplate reader after 24 h for bacteria and after 48 h for fungi. In each case, triplicate tests were performed and the average was taken as final reading. MIC was expressed as the lowest concentration inhibiting test organism’s growth.54)
Antimycobacterial ActivityM. tuberculosis (RCMB 010126) strain was provided from culture collection of the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Cairo, Egypt. The isolated M. tuberculosis (RCMB 010126) clone was cultivated under agitation on LB medium at 37°C for 24 h. The antitubercular activity was expressed as the diameter of inhibition zones using agar well diffusion method and as MIC using serial dilution technique. Isoniazide and pyrazinamide were used as the reference drugs. The final solutions concentrations were 125, 62.50, 31.25, 15.63, 7.81, 3.90, 1.95, 0.98, 0.49, 0.24 and 0.12 µmol/mL. The zones of inhibition were analyzed after 72 h of incubation at 37°C. Each test was repeated 3 times. MIC was expressed as the lowest concentration inhibiting test organism’s growth.
In Vitro CytotoxicityMDA-MB231 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM)/high glucose supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 1% penicillin/streptomycin. The new compounds were evaluated in a primary five cell line-one concentration (25 mg/mL) anticancer assay against MDA-MB231 cells cell lines. The cytotoxic effect of the newly synthesized compounds was evaluated by testing the capacity of the reducing enzymes present in viable cells to convert MTT to formazan crystals as previously described,55) with some modifications. Briefly, cells cultured in complete medium were seeded into 96-well microtiter plates (in quintuplicates) with 2×104 cells per well and incubated at 37°C under a humidified atmosphere of 5% CO2 for 24 h. The cell medium in test wells were then changed to serum free medium (SFM) containing 25 mg/mL of the test compounds, while the cell medium in control wells were changed to SFM containing an equivalent volume of solvent (dimethyl sulfoxide “DMSO”). After incubation at 37°C for 24 h, SFM in control and test wells were replaced by 100 mL/well of MTT; 0.5 mg/mL) in phosphate-buffered saline (PBS) and incubated at 37°C for an additional 3 h. MTT solution were removed and the purple formazan crystals formed at the bottom of the wells were dissolved using 100 mL isopropyl alcohol/well with shaking for 1 h at room temperature. The absorbance at 549 nm was read on a microplate reader (ELX 800; Bio-Tek Instruments, Winooski, VT, U.S.A.). The dose response curves of the compounds effecting >50% inhibition in one-dose prescreening for each cell line were established with concentrations of 25, 12.5, 6.25, 3.125, 1.56 and 0.78 mg/ mL, and the concentrations causing 50% cell growth inhibition (IC50) were calculated.
Molecular DockingThe molecular docking of the tested compounds was performed using Discovery Studio 4/CDOCKER protocol (Accelrys Software Inc.). The protein crystallographic structure of Bacillus anthracis dihydropteroate synthase (PDB code 3TYE) was downloaded from the Protein Data Bank (PDB). The protein was prepared for docking process according to the standard protein preparation procedure integrated in Accelry’s discovery studio 4 and prepared by prepare protein protocol. Sulfathiazole-6-hydroxymethyl-7,8-dihydropterin-pyrophosphate (STZ-DHPP) adduct and the 21f were drawn as a database and prepared by prepare ligand protocol to generate 3D structure and refine using CHARMM force field with full potential. Docking simulations were run using CDOCKER protocol where a maximum bad orientations was 800 and orientation van der Waals (vdW) energy threshold was 300. Simulated annealing simulation would be then carried out consisting of a heating phase 700 K with 2000 steps and a cooling phase back to 5000 steps. The binding energy was calculated as a score to rank the docking poses. The top 10 docking poses would be finally saved. Docking poses were ranked according to their –CDOCKER interaction energy, and the top poses were chosen for analysis of interactions for each compound.