Abstract
The direct inhibitory potential of twenty five anti-tuberculosis drugs on eight CYP-specific reactions in human liver microsomes was investigated to predict in vivo drug–drug interactions (DDIs) from in vitro data. Rifampicin, rifabutin, and thioacetazone inhibited one CYP reaction. Isoniazid and clofazimine had inhibitory effects on four CYP reactions, and rifapentine, ethionamide, and prothionamide widely inhibited CYP reactions. Based on the inhibition constant (Ki) and the therapeutic total inhibitor concentrations [I]max of eight drugs in human plasma, [I]max/Ki values were calculated to evaluate clinical DDIs. The [I]max/Ki values were 0.20 or less for rifampicin, rifabutin, and thioacetazone; 0.15–2.0 for isoniazid; 0.14–1.5 for rifapentine; 0.29–1.4 for ethionamide; 0.41–2.2 for prothionamide; and 0.12–6.3 for clofazimine. The highest [I]max/Ki values were 2.0 for isoniazid on CYP3A4 [testosterone (T)]; 1.5 for rifapentine on CYP3A4 [midazolam (M)]; 1.4 for ethionamide on CYP2C8; 2.2, 1.8, and 1.3 for prothionamide on CYP2B6, CYP2C19, and CYP2C8, respectively; and 6.3 and 5.7 for clofazimine on CYP3A4 (M) and CYP3A4 (T), respectively. These drugs with high [I]max/Ki values lead to clinical DDIs. Considering the drug regimens for tuberculosis (TB) and co-infection with TB and human immunodeficiency virus, the inhibitory potential for CYP3A4 and CYP2B6 is particularly important. These results suggest that clofazimine and prothionamide are likely to cause clinically relevant DDIs when co-administered with products metabolized by CYP3A4 and CYP2B6, respectively. Isoniazid and rifapentine may cause DDIs with drugs metabolized by CYP3A4.
Tuberculosis (TB) is a global health problem. In 2013, an estimated 9.0 million people developed TB and 1.5 million died from the disease, 360000 of whom were human immunodeficiency virus (HIV)-positive.1) Multidrug-resistant TB (MDR-TB) is defined as TB with resistance to at least isoniazid and rifampicin, the two most powerful first-line anti-TB drugs.2) Current treatment regimens for MDR-TB are more toxic, last longer, and are less effective than treatment regimens for drug-sensitive TB.3) In the development of combination regimens for TB and co-infection with TB and HIV, the prediction of drug–drug interactions (DDIs) helps to avoid the risk of adverse reactions caused by DDIs related to CYP enzymes.
It is estimated that CYP enzymes are responsible for the metabolism of approximately three-quarters of the most prescribed drugs that are cleared by metabolism, including the anti-retroviral agents used to treat HIV.4,5) Several CYP enzymes are principally involved in the oxidation of a large number of xenobiotic chemicals as well as endogenous compounds. Their expression has been reported at the protein level in human liver microsomes.6) CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 [midazolam (M) and testosterone (T)] are reported to be important enzymes in the guidelines on the investigation of DDIs published by the U.S. Food and Drug Administration7) and the European Medicines Agency.8)
It has been reported that isoniazid coadministration increased the total area under the concentration–time curve (AUC) of triazolam (substrate of CYP3A4) after a single oral dose.9) Horita and Doi10) recently reported the effects of anti-TB drugs on CYP3A4 activity using a commercially available screening kit for CYP3A4 inhibitors. However, information on the CYP inhibitory potential for anti-TB drugs both in vivo and in vitro is limited. It is therefore critical to understand the ability of anti-TB drugs to inhibit CYP enzymes. In the present study, twenty five anti-TB drugs were selected by reference to a World Health Organization (WHO) guideline published in 2011.11) To predict DDIs, the direct inhibitory effects of these anti-TB drugs on eight substrate reactions for seven CYP enzymes were evaluated using human liver microsomes in vitro.
MATERIALS AND METHODS
Chemicals and ReagentsIsoniazid, rifampicin, ethambutol dihydrochloride, pyrazinamide, rifabutin, levofloxacin, ofloxacin, ethionamide, D-cycloserine, p-aminosalicylic acid, clofazimine, linezolid, potassium clavulanate, clarithromycin, and imipenem monohydrate were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Rifapentine, moxifloxacin hydrochloride, capreomycin sulfate, and thioacetazone were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Streptomycin sulfate, kanamycin A, amikacin disulfate, gatifloxacin, and prothionamide were purchased from LKT Laboratories (Saint Paul, MN, U.S.A.). Amoxicillin trihydrate was purchased from Tokyo Chemical Industry (Tokyo, Japan). CYP enzyme-specific marker substrates, metabolites, positive controls, and internal standards were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan), Sigma-Aldrich, Toronto Research Chemicals (Toronto, Canada), Corning (Woburn, MA, U.S.A.), Tokyo Chemical Industry, Santa Cruz Biotechnology, and Alsachim (Strasbourg, France). Reduced nicotinamide adenine dinucleotide phosphate (NADPH) and reduced nicotinamide adenine dinucleotide (NADH) were purchased from Oriental Yeast (Tokyo, Japan). Pooled human liver microsomes (150 donors) were supplied by Corning. All other chemicals and solvents were of the highest chemical grade available.
Incubation ConditionsThe standard incubation mixture consisted of 100 mM potassium phosphate buffer (pH 7.4), 2.5 mM NADPH/NADH mixture solution, microsomes, a substrate, and a test compound or a positive control inhibitor. The experimental conditions for CYP inhibitory assays are summarized in Table 1. Substrate was incubated at a concentration equal to previously determined Michaelis–Menten constant (Km) value. The substrates and inhibitors were dissolved in acetonitrile–dimethyl sulfoxide (DMSO) (9 : 1, v/v) or methanol (bupropion). The test compounds at seven concentrations were prepared in DMSO, acetonitrile–DMSO (9 : 1, v/v), or saline. The final concentration of organic solvents in each reaction mixture was 1% (v/v) or less. The highest concentration of test compounds was set at equal to or more than the maximum concentration (Cmax) in human plasma. After preincubation at 37°C for 5 min, the reaction was initiated by adding NADPH/NADH solution in a final volume of 400 µL in duplicate. After the incubation, the reaction was terminated with 400 µL of ice-cold acetonitrile–methanol (1 : 1, v/v) containing each internal standard. Those samples and standard curve samples were centrifuged at 5013×g for 10 min, and the supernatants were subjected to HPLC separation with LC-MS/MS.
Table 1. Summary of Experimental Conditions for CYP Inhibitory Assays
| CYP enzyme | Reaction | Substrate | (µM) | Incubation time (min) | Protein (mg/mL) | Positive control | (µM) |
|---|
| CYP1A2 | Phenacetin O-deethylation | Phenacetin | 50 | 20 | 0.1 | α-Naphthoflavone | 1 |
| CYP2B6 | Bupropion hydroxylation | Bupropion | 150 | 20 | 0.1 | Sertraline | 50 |
| CYP2C8 | Paclitaxel 6α-hydroxylation | Paclitaxel | 10 | 20 | 0.1 | Quercetin | 50 |
| CYP2C9 | Diclofenac 4′-hydroxylation | Diclofenac | 10 | 10 | 0.1 | Sulfaphenazole | 10 |
| CYP2C19 | S-Mephenytoin 4′-hydroxylation | S-Mephenytoin | 30 | 30 | 0.2 | Tranylcypromine | 50 |
| CYP2D6 | Bufuralol 1′-hydroxylation | Bufuralol | 10 | 10 | 0.1 | Quinidine | 10 |
| CYP3A4 (Ma)) | Midazolam 1′-hydroxylation | Midazolam | 2 | 10 | 0.1 | Ketoconazole | 10 |
| CYP3A4 (Tb)) | Testosterone 6β-hydroxylation | Testosterone | 100 | 10 | 0.1 | Ketoconazole | 10 |
a) Midazolam. b) Testosterone.
All measurements were conducted using an API4000 or 4000QTRAP mass spectrometer (AB Sciex, Foster City, CA, U.S.A.) and a Prominence HPLC system (Shimadzu, Kyoto, Japan). The measurement by LC-MS/MS was operated in the multiple reaction monitoring (MRM) mode and the positive mode for electrospray ionization. The metabolite used in the analytical method was acetaminophen for CYP1A2; hydroxybupropion for CYP2B6; 6α-hydroxypaclitaxel for CYP2C8; 4′-hydroxydiclofenac for CYP2C9; 4′-hydroxymephenytoin for CYP2C19; 1′-hydroxybufuralol for CYP2D6; 1′-hydroxymidazolam for CYP3A4 (M), and 6β-hydroxytestosterone for CYP3A4 (T). Each stable isotope was used as the internal standard. HPLC separation was performed using a Cadenza CD-C18 (2.0×100 mm, 3 µm; Imtakt, Kyoto, Japan) and with mobile phase A [1 mM ammonium formate aqueous–formic acid (1000 : 2, v/v)] and B (methanol). The gradient program was 10 (0.0–1.0)→90 (5.0–9.0)→10 (9.1–15.0) (%B (min)), and the flow rate was 0.25 mL/min. For all measurements, the column oven temperature was set at 40°C.
Data AnalysisAll inhibition data were calculated using the mean of data in duplicate. The IC50 values were calculated using Phoenix WinNonlin version 6.3 (Pharsight, St. Louis, MO, U.S.A.). The inhibition constant (Ki) was calculated using the equation Ki=0.5×IC50 by assuming competitive inhibition in all cases, as substrate was investigated at a concentration equal to previously determined Km value. The therapeutic total inhibitor concentration [I]max, which is the Cmax, in human plasma was obtained from pharmaceutical package inserts, the Handbook of Anti-tuberculosis Agents,12) and a previously published report.13) [I]max/Ki values were calculated from Ki and reported [I]max values. Further, unbound plasma concentration ([I]max,u) and total and unbound hepatic input concentrations ([I]in and [I]in,u) were determined for CYP3A4 based on reports by Ito et al.14) and Obach et al.15) The unbound fraction (fu) of anti-TB drugs was obtained from a previously published report.16)
RESULTS AND DISCUSSION
According to the WHO guideline published in 2011,11) the anti-TB drugs are divided into five groups: first-line drugs, second-line parenteral drugs, fluoroquinolones, oral bacteriostatic second-line drugs, and other group 5 drugs. The choice of drug depends on the drug-susceptibility test or close contacts with MDR-TB, previous use of the drug in the patient, and the frequency of its use or documented background drug resistance in the setting. However, information on the CYP inhibitory potential for anti-TB drugs both in vivo and in vitro is limited. With this background, the potential for direct inhibition of twenty five anti-TB drugs on eight CYP specific reactions was investigated using human liver microsomes in vitro, and [I]max/Ki values were calculated to evaluate risks for clinical DDIs.
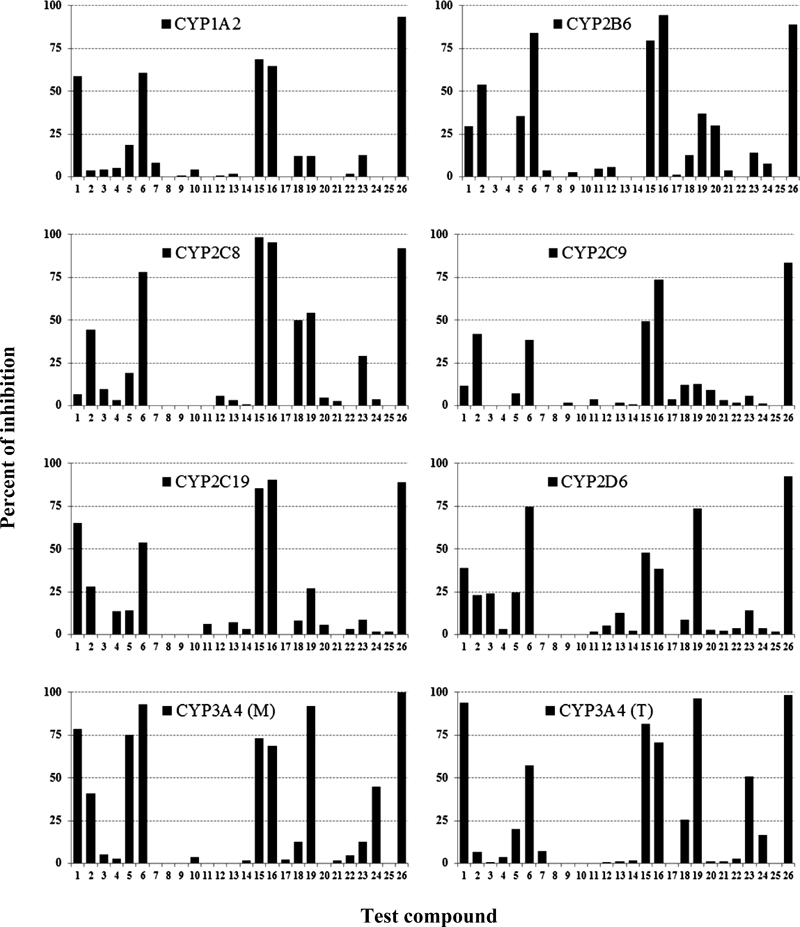
The percent of inhibition of anti-TB drugs at the highest concentration used on typical CYP reactions is shown in Fig. 1. The IC50 and [I]max/Ki values are shown in Tables 2 and 3, respectively. Of the drugs investigated in the present study, eight drugs inhibited one or more CYP reactions. The other seventeen drugs showed no IC50 values for all eight CYP reactions.
Table 2. IC
50 Value of Anti-tuberculosis Drugs on Typical CYP Reactions
| Test compound | Concentration (µM) | IC50 (µM) |
|---|
| CYP1A2 | CYP2B6 | CYP2C8 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 (M) | CYP3A4 (T) |
|---|
| Isoniazid | 1–1000 | 773 | — | — | — | 605 | — | 285 | 58.5 |
| Rifampicin | 0.3–300 | — | 237 | — | — | — | — | — | — |
| Rifabutin | 0.03–30 | — | — | — | — | — | — | 8.55 | — |
| Rifapentine | 0.3–300 | 246 | 64.2 | 115 | — | 214 | 81.6 | 23.0 | 232 |
| Ethionamide | 1–1000 | 524 | 396 | 110 | — | 195 | — | 451 | 282 |
| Prothionamide | 0.3–300 | 188 | 34.3 | 57.6 | 153 | 43.6 | — | 180 | 148 |
| Clofazimine | 0.03–30 | — | — | 14.1 | — | — | 4.54 | 0.275 | 0.304 |
| Thioacetazone | 0.1–100 | — | — | — | — | — | — | — | 98.0 |
Table 3. Assessment of Drug–Drug Interactions of Anti-tuberculosis Drugs on Typical CYP Reactions
| Test compound | Dose (mg) | [I]maxa) | [I]max/Kib) |
|---|
| (µg/mL) | (µM) | CYP1A2 | CYP2B6 | CYP2C8 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 (M) | CYP3A4 (T) |
|---|
| Isoniazid | 300 | 8.00 | 58.4 | 0.15 | — | — | — | 0.19 | — | 0.41 | 2.0 |
| Rifampicin | 450 | 7.99 | 9.71 | — | 0.082 | — | — | — | — | — | — |
| Rifabutin | 600 | 0.724 | 0.855 | — | — | — | — | — | — | 0.20 | — |
| Rifapentine | 600 | 15.1 | 17.2 | 0.14 | 0.53 | 0.30 | — | 0.16 | 0.42 | 1.5 | 0.15 |
| Ethionamide | 500 | 12.5 | 75.2 | 0.29 | 0.38 | 1.4 | — | 0.77 | — | 0.33 | 0.53 |
| Prothionamide | 250 | 6.94 | 38.5 | 0.41 | 2.2 | 1.3 | 0.50 | 1.8 | — | 0.43 | 0.52 |
| Clofazimine | 200 | 0.408 | 0.862 | — | — | 0.12 | — | — | 0.38 | 6.3 | 5.7 |
| Thioacetazone | 150 | 1.59 | 6.73 | — | — | — | — | — | — | — | 0.14 |
a) Total inhibitor concentration ([I]max), which is the Cmax, was obtained from pharmaceutical package inserts, the Handbook of Anti-tuberculosis Agents,12) and a previously published report.13) b) Inhibition constant (Ki) was calculated using the equation, Ki=0.5×IC50 by assuming competitive inhibition in all cases, as substrate was investigated at a concentration equal to previously determined Michaelis–Menten value. — Not calculated.
The first-line drugs rifampicin and rifabutin inhibited CYP2B6 and CYP3A4 (M), respectively; however, the [I]max/Ki values were 0.20 or less. Isoniazid had inhibitory effects on four CYP reactions. The [I]max/Ki values for isoniazid on CYP1A2, CYP2C19, and CYP3A4 (M) were 0.41 or less. The highest [I]max/Ki value was 2.0 for CYP3A4 (T). Rifapentine widely inhibited CYP reactions. The [I]max/Ki values for rifapentine on CYP1A2, CYP2B6, CYP2C8, CYP2C19, CYP2D6, and CYP3A4 (T) were 0.53 or less, and the highest [I]max/Ki value was 1.5 for CYP3A4 (M). These results suggest that isoniazid and rifapentine may cause DDIs with CYP3A4. The difference in IC50 values between midazolam- and testosterone-mediated reactions is considered to reflect the difference in the CYP3A4-binding sites. Based on chemical inhibition characterizations and substrate correlation analyses, midazolam and testosterone seem to belong to two distinct groups of CYP3A4 substrates.17)
The second-line injectable drugs kanamycin, amikacin, and capreomycin showed no inhibition on eight CYP reactions, and the fluoroquinolones levofloxacin, moxifloxacin, gatifloxacin, and ofloxacin also showed no inhibition on eight CYP reactions. These second-line injectable drugs and fluoroquinolones are often used to treat MDR-TB patients, and the results indicate that inhibition on CYP reactions by these drugs may not need to be taken into account in combination treatment for MDR-TB patients.
The orally administrated second-line drugs ethionamide and prothionamide widely inhibited the same CYP reactions. The [I]max/Ki values of ethionamide on CYP1A2, CYP2B6, CYP2C19, CYP3A4 (M), and CYP3A4 (T) were 0.77 or less, and those of prothionamide on CYP1A2, CYP2C9, CYP3A4 (M), and CYP3A4 (T) were 0.52 or less. The highest [I]max/Ki value for ethionamide was 1.4 on CYP2C8, and the highest [I]max/Ki values for prothionamide were 2.2, 1.8, and 1.3 on CYP2B6, CYP2C19, and CYP2C8, respectively. These inhibitory results suggest that prothionamide is likely to cause clinically relevant DDIs with CYP2B6. The inhibition of CYP2C19 and CYP2C8 by prothionamide and that of CYP2C8 by ethionamide may cause DDIs. Ethionamide and prothionamide have a pyridine moiety in their chemical structure, as does isoniazid. Uncoupled pairs of nitrogen atoms in pyridine are considered to inhibit CYP.18)
Of the group 5 drugs, thioacetazone inhibited CYP3A4 (T) reaction; however, the [I]max/Ki value was only 0.14. Clofazimine had inhibitory effects on four CYP reactions. The [I]max/Ki values for clofazimine were 0.38 on CYP2D6 and 0.12 on CYP2C8, and the highest [I]max/Ki values were 6.3 on CYP3A4 (M) and 5.7 on CYP3A4 (T). Horita and Doi10) reported that the IC50 value of clofazimine on CYP3A4-mediated 7-benzyloxy-trifluoromethylcoumarin metabolism was nearly equal to the Cmax, suggesting that clofazimine has an [I]max/Ki value of 1 or higher.
Further, total and unbound plasma concentrations ([I]max and [I]max,u) and hepatic concentrations ([I]in and [I]in,u) for CYP3A4 were calculated and [I]/Ki values were compared. These results are shown in Table 4. AUC ratios (1+[I]/Ki values) for isoniazid calculated by [I]max and [I]max,u were from 1.41 to 3.0, and those by [I]in and [I]in,u were from 2.3 to 7.6. It is reported that isoniazid coadministration increased the total AUC of triazolam (substrate of CYP3A4) after a single oral dose by 1.5 folds (control (26.5 ng·h/mL) vs. with isoniazid (38.6 ng·h/mL)).9) The use of total or unbound plasma concentration may be an adequate method because [I]max/Ki and [I]max,u/Ki values of isoniazid were identical with the AUC ratio. As clofazimine shows high [I]/Ki values by four methods, drugs metabolized by CYP3A4 should be carefully administered with clofazimine.
Table 4. Assessment of Drug–Drug Interactions of Anti-tuberculosis Drugs on CYP3A4
| Test compound | fua) | CYP3A4 (M) | CYP3A4 (T) |
|---|
| [I]max/Ki | [I]max,u/Kib) | [I]in/Kic) | [I]in,u/Kid) | [I]max/Ki | [I]max,u/Kib) | [I]in/Kic) | [I]in,u/Kid) |
|---|
| Isoniazid | 0.99 | 0.41 | 0.41 | 1.4 | 1.3 | 2.0 | 2.0 | 6.6 | 6.6 |
| Rifampicin | 0.10 | — | — | — | — | — | — | — | — |
| Rifabutin | 0.15 | 0.20 | 0.030 | 10 | 1.6 | — | — | — | — |
| Rifapentine | 0.03 | 1.5 | 0.045 | 5.2 | 0.16 | 0.15 | 0.0044 | 0.51 | 0.015 |
| Ethionamide | 0.70 | 0.33 | 0.23 | 1.2 | 0.81 | 0.53 | 0.37 | 1.9 | 1.3 |
| Prothionamide | 0.40 | 0.43 | 0.17 | 1.4 | 0.55 | 0.52 | 0.21 | 1.7 | 0.67 |
| Clofazimine | 0.30 | 6.3 | 1.9 | 197 | 59 | 5.7 | 1.7 | 178 | 53 |
| Thioacetazone | 0.05 | — | — | — | — | 0.14 | 0.0069 | 0.94 | 0.047 |
a) The unbound fraction (fu) of anti-TB drugs was obtained from a previously published report.16) b) [I]max,u was calculated from [I]max×fu. c) [I]in was calculated from the equation, [I]max+kaFaD/Qh. The values of ka (absorption rate constant), Fa (fraction absorbed from gut into portal vein), Qh (hepatic blood flow rate), and RB (blood-to-plasma concentration ratio) were assumed to be 0.1 min−1, 1, 1610 mL min−1, and 1, respectively, and D was dose. d) [I]in,u was calculated from [I]in×fu. — Not calculated.
Of novel drugs and candidates currently undergoing clinical trials, bedaquiline (TMC-207), pretomanid (PA-824), and sutezolid have been reported to be substrates of CYP3A4.19) In addition, HIV protease inhibitors are substrates of CYP3A4.20) Efavirenz, a non-nucleoside reverse transcriptase inhibitor, is a substrate of CYP3A4 and CYP2B6.20,21) Considering the drug regimens for TB and co-infection with TB and HIV, the potential for CYP3A4 and CYP2B6 inhibition is particularly important. In conclusion, clofazimine and prothionamide are likely to cause clinically relevant DDIs when co-administered with products that are metabolized by CYP3A4 and CYP2B6, respectively. Isoniazid and rifapentine may cause DDIs via the inhibition of CYP3A4.
Acknowledgment
We are grateful to Dr. Hiroyuki Sasabe of Tokushima Research Institute, Otsuka Pharmaceutical Co., Ltd. for helpful advice with scientific discussion.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) World Health Organization. “Global tuberculosis report 2014. WHO/HTM/TB/2014.08.”: ‹http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf›
- 2) Chang KC, Yew WW. Management of difficult multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis: update 2012. Respirology, 18, 8–21 (2013).
- 3) Falzon D, Jaramillo E, Schünemann HJ, Arentz M, Bauer M, Bayona J, Blanc L, Caminero JA, Daley CL, Duncombe C, Fitzpatrick C, Gebhard A, Getahun H, Henkens M, Holtz TH, Keravec J, Keshavjee S, Khan AJ, Kulier R, Leimane V, Lienhardt C, Lu C, Mariandyshev A, Migliori GB, Mirzayev F, Mitnick CD, Nunn P, Nwagboniwe G, Oxlade O, Palmero D, Pavlinac P, Quelapio MI, Raviglione MC, Rich ML, Royce S, Rüsch-Gerdes S, Salakaia A, Sarin R, Sculier D, Varaine F, Vitoria M, Walson JL, Wares F, Weyer K, White RA, Zignol M. WHO guidelines for the programmatic management of drug-resistant tuberculosis: 2011 update. Eur. Respir. J., 38, 516–528 (2011).
- 4) Guengerich FP, Rendic S. Update information on drug metabolism systems—2009, Part I. Curr. Drug Metab., 11, 1–3 (2010).
- 5) Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov., 4, 825–833 (2005).
- 6) Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther., 270, 414–423 (1994).
- 7) U.S. Food and Drug Administration. (FDA). “Guidance for industry: Drug interaction studies—study design, data analysis, implication for dosing, and labeling recommendations. Draft guidance. February 2012.”: ‹http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf›
- 8) European Medicines Agency. “Guideline on the investigation of drug interactions. CPMP/EWP/560/95/Rev.1 Corr.* (2012).”: ‹http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf›
- 9) Ochs HR, Greenblatt DJ, Knüchel M. Differential effect of isoniazid on triazolam oxidation and oxazepam conjugation. Br. J. Clin. Pharmacol., 16, 743–746 (1983).
- 10) Horita Y, Doi N. Comparative study of the effects of antituberculosis drugs and antiretroviral drugs on cytochrome P450 3A4 and P-glycoprotein. Antimicrob. Agents Chemother., 58, 3168–3176 (2014).
- 11) World Health Organization. “Guidelines for the programmatic management of drug-resistant tuberculosis, 2011 update. WHO/HTM/TB/2011.6.”: ‹http://whqlibdoc.who.int/publications/2011/9789241501583_eng.pdf›
- 12) Global Alliance for Drug Development TB. Handbook of antituberculosis agents. Introduction. Tuberculosis, 88: 85–86 (2008). ‹http://dx.doi.org/10.1016/S1472-9792(08)70002-7›
- 13) Peloquin CA, Nitta AT, Berning SE, Iseman MD, James GT. Pharmacokinetic evaluation of thiacetazone. Pharmacotherapy, 16, 735–741 (1996).
- 14) Ito K, Brown HS, Houston JB. Database analyses for the prediction of in vivo drug–drug interactions from in vitro data. Br. J. Clin. Pharmacol., 57, 473–486 (2004).
- 15) Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM. The utility of in vitro cytochrome P450 inhibition data in the prediction of drug–drug interactions. J. Pharmacol. Exp. Ther., 316, 336–348 (2006).
- 16) Lakshminarayana SB, Huat TB, Ho PC, Manjunatha UH, Dartois V, Dick T, Rao SP. Comprehensive physicochemical, pharmacokinetic and activity profiling of anti-TB agents. J. Antimicrob. Chemother., 70, 857–867 (2015).
- 17) Kenworthy KE, Bloomer JC, Clarke SE, Houston JB. CYP3A4 drug interactions: correlation of 10 in vitro probe substrates. Br. J. Clin. Pharmacol., 48, 716–727 (1999).
- 18) Lesca P, Lecointe P, Paoletti C, Mansuy D. Ellipticines as potent inhibitors of aryl hydrocarbon hydroxylase: their binding to microsomal cytochromes P450 and protective effect against benzo(a)pyrene mutagenicity. Biochem. Pharmacol., 27, 1203–1209 (1978).
- 19) Dooley KE, Kim PS, Williams SD, Hafner RTB. TB and HIV Therapeutics: Pharmacology Research Priorities. Aids Res. Treat., 2012, 874083 (2012).
- 20) Barry M, Mulcahy F, Merry C, Gibbons S, Back D. Pharmacokinetics and potential interactions amongst antiretroviral agents used to treat patients with HIV infection. Clin. Pharmacokinet., 36, 289–304 (1999).
- 21) Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther., 306, 287–300 (2003).