Abstract
RNA interference via small interfering RNA (siRNA) has many potential therapeutic applications, and liposomal-based systems are useful for improving the pharmacokinetics of siRNAs, including their intracellular release and distribution. However, for the successful translation of this technology into clinical applications, it is important to understand how liposomal encapsulation changes the cellular uptake and immunostimulatory adverse effects of siRNAs. Here we evaluated the cellular uptake and innate immune activation by an immunostimulatory siRNA encapsulated within a liposome carrier in commercially available human peripheral blood mononuclear cells (PBMCs). We found considerable lot-to-lot variation in cytokine production by the PBMCs. Flow cytometric analysis in conjunction with intracellular staining of tumor necrosis factor-α (TNF-α) revealed that after treating PBMCs with the liposomal siRNA, approximately 5% of the cells produced TNF-α and more than 90% of the TNF-α-producing cells were positive for CD14 expression. We also showed that peripheral blood CD14+ monocytes in the cytokine release assay had low inter-lot variabilities in TNF-α production, suggesting that the peripheral blood CD14+ monocyte-based cytokine release assay is a specific means of alleviating the lot-to-lot variability in the cytokine release profiles of commercially available PBMCs. Our results also show that the peripheral blood CD14+ monocyte-based cytokine release assay can be used to identify the siRNA recognition receptors that mediate individual cytokine production.
Small interfering RNA (siRNA) is a class of double-stranded RNA, generally 19 to 23 base pairs in length with two nucleotides overhanging the 3′ end, that can be used for sequence-specific gene silencing of mRNA.1) RNA interference using siRNA is a powerful tool for gene function studies, and the hope now is to develop this technology into therapeutic applications.2–4) The potency and specificity of naked siRNA make it suitable for studying genes in vitro; however, improvements to this technology are needed before it can be used therapeutically in vivo. For example, naked siRNAs are rapidly degraded by nucleases in the blood and they cannot penetrate cell membranes because of their large molecular size (approx. 13 kDa) and high hydrophilicity (approx. 40 negatively charged phosphate groups).5,6) To improve the pharmacokinetic profile of siRNAs, a variety of drug delivery carriers have been developed, such as liposomes and polymeric micelles.3,7) Other strategies have also been investigated to increase the stability of siRNA in serum, such as the use of chemically modified RNA containing 2′-O-methyl (2′-OMe) or 2′-fluoro modified nucleotides.8)
Although several studies have demonstrated efficient delivery to target organs or tissues and beneficial therapeutic effects of siRNAs in vivo, it has also become clear that siRNAs have the potential to induce unwanted immune responses, including excessive cytokine release.9–11) Kleinman et al. demonstrated that investigational siRNAs induced sequence-nonspecific inhibition of angiogenesis via the activation of toll-like receptor 3 (TLR3), which is located in the lipid bilayer of plasma membrane and early, late, and recycling endosomes.11,12) Kleinman et al. further showed that unmodified 21-mer siRNA and 2′-OMe-siRNA both activate TLR3.11) Other groups have reported that siRNA containing immunostimulatory motifs such as 5′-UGU-3′ powerfully induce immune cell-mediated tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6), and interferon-α production.13,14) Although the exact sequence dependence and mechanisms for siRNA-mediated immune activation are not fully understood, it appears that TLR3,11,15) TLR7,16) and TLR817) are involved in the recognition of siRNAs, leading to cytokine production. TLR8 is phylogenetically and structurally similar to TLR7, however, there is species-specific difference in single-stranded RNA recognition.18–20) For example, the imidazoquinoline resiquimod activated human TLR7, TLR8 and murine TLR7, but not murine TLR8.21) The ability of the immune system to distinguish between self and non-self RNA (i.e., viral RNA) is key to reducing the TLR-mediated innate immune response to siRNA. Experimental evidence suggests that mimicking self-RNA through nucleoside modification of siRNA is one means of reducing the TLR-mediated innate immune response to siRNA.22–24) However, depending on the extent and location of these modifications, they may not only prevent the unwanted induction of cytokines but may also block the wanted gene-silencing activity of the siRNA.16)
To decrease the immunostimulatory toxicity of siRNA, nanotechnology-based carriers can be used to control the biodistribution, intracellular behavior, and release of siRNA at specific cellular organelles to avoid recognition by TLRs.25,26) Cell-type-specific uptake of nanoparticles by peripheral blood mononuclear cells (PBMCs) and the resultant cellular responses have been reported previously.27) It has also been reported that certain nanoparticles themselves have immunostimulatory effects.28) Therefore, to avoid exaggerating the undesirable immunostimulatory effects of siRNA through encapsulation within nanosized carriers, it is important to evaluate the immunological adverse effects of nanotechnology-based therapeutics as complete products. Furthermore, because of species-specific differences in immunological responses to potential therapeutic agents, in the preclinical setting it is useful to evaluate their immunotoxicity in human whole blood or human-derived cell lines in vitro.29,30) The majority of PBMCs are lymphocytes (e.g., T cells) and monocytes; so, PBMCs represent a well-defined subpopulation of host defense cells that can be used to examine the release of endogenous mediators induced by biopharmaceuticals such as pro-inflammatory cytokines.31)
In the present study, we encapsulated a known immunostimulatory siRNA within a liposomal carrier and evaluated both the cellular uptake of the siRNA by commercially available PBMCs and the resultant innate immune response. We also examined how to reduce the lot-to-lot variability in the cytokine release profiles of commercially available PBMCs by examining the cell-specific responses to our liposomal siRNA and by identifying the siRNA recognition receptor.
MATERIALS AND METHODS
Cell Culture and ReagentsHuman PBMCs (Cellular Technology Ltd., Shaker Heights, OH, U.S.A.) and human peripheral blood CD14+ monocytes (Lonza, Walkersville, MD, U.S.A.) from healthy donors were cultured in RPMI-1640 medium supplemented with 2 mM GlutaMax (Thermo Fisher Scientific, Waltham, MA, U.S.A.) and 10% heat-inactivated fetal bovine serum (FBS). Cells were grown in a humidified incubator at 37°C under an atmosphere of 5% CO2. 2,3-Dioleoyl-N,N,N-trimethylammnomiopropane (DOTAP) was purchased from Avanti Polar Lipids (Alabaster, AL, U.S.A.). N-[(Methoxy polyethyleneglycol 2000)(ylcarbonyl)]-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt (PEG-DSPE) and egg phosphatidylcholine were purchased from NOF corporation (Tokyo, Japan). Double-stranded siRNA and siRNA labeled with Alexa647 at the 5′ end of the sense strand (A647-siRNA) were chemically synthesized by Gene Design Inc. (Osaka, Japan). A previously reported immunostimulatory siRNA with 2-nt 3′-overhangs targeting β-galactosidase (β-gal 728) was used (sense, 5′-CUA CAC AAA UCA GCG AUU UdTdT-3′; anti-sense, 5′-AAA UCG CUG AUU UGU GUA GdTdT-3′).13) Polyinosinic : polycytidylic acid (poly(I : C)) (TLR3 agonist) and CL097 (TLR7/8 agonist) were obtained from Invitrogen (San Diego, CA, U.S.A.) and resuspended in endotoxin-free sterile water and stored at −20°C until use. For immunostaining, human Fc Block, 7-aminoactinomycin D, protein transport inhibitor (containing brefeldin A), and Cytofix/Cytoperm solution were purchased from BD Biosciences (Franklin Lakes, NJ, U.S.A.). Fluorescein isothiocyanate- or phycoerythrin-conjugated antibodies against CD14, TNF-α, and the respective mouse isotype controls were purchased from BD Biosciences. For the TLR inhibition assay, ODN2087 (TLR7/8 antagonists) and ODN20958 (TLR7 antagonist) were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany), and CU-CPT4a (TLR3 antagonist) was purchased from R&D Systems (Minneapolis, MN, U.S.A.).
Preparation of LiposomesLiposomes were prepared by using the ethanol dilution method previously reported.32–34) Briefly, DOTAP and PEG-DSPE were mixed and hydrated in distilled water at a molar ratio of 10 : 1. After hydration, the dispersion was sonicated for 2 min with a tip-type sonicator (Sonics & Materials, Newtown, CT, U.S.A.) to make a cationic lipid core. The cationic lipid core (47 mM) was mixed with siRNA (24 mg/mL) at a nitrogen/phosphate ratio of 2.26 in distilled water. To encapsulate the core, the cationic lipid core/siRNA complex was then diluted with 62.5% (v/v) ethanol containing 28 mM total lipid of egg phosphatidylcholine and PEG-DSPE (9 : 1). Distilled water was added to the solution to dilute the ethanol to 5% (v/v). To remove the ethanol, the diluted solution was concentrated by means of ultrafiltration in Vivaspin spin columns (molecular weight cut-off, 100 kDa; GE Healthcare, Buckinghamshire, U.K.), after which the liposomal solution remaining in the upper column was diluted with phosphate-buffered saline. The size distribution and zeta potential of the liposomes were determined with a dynamic light-scattering spectrophotometer (Zetasizer Nano; Malvern Instruments, Worcestershire, U.K.). The degree of siRNA encapsulation was determined by quantifying the amounts of unencapsulated siRNA and total siRNA using membrane-impermeable RiboGreen (Thermo Fisher Scientific) before and after the addition of 0.1% Triton-X100, respectively, to disrupt the lipid bilayers. The efficiency of siRNA encapsulation (%) was calculated as [(total siRNA amount − unloaded siRNA amount)/(total siRNA amount)]×100.
siRNA Stability AssayTo evaluate the stability of our liposomal siRNA in the presence of a nuclease, 1 µg of naked siRNA, siRNA/cationic core complex, or liposomal siRNA was incubated with 80 ng of RNase A (Qiagen, Hilden, Germany) at 37°C for 0.5, 1, 2, 4, 8 or 24 h, respectively. The mixtures were added directly to gel loading buffer containing 0.1% sodium dodecyl sulfate. The siRNA were run on a 15% polyacrylamide gel and stained with SYBR Safe (Thermo Fisher Scientific) staining.
Stimulation of CellsHuman PBMCs and CD14+ monocytes (purity, >95%) were seeded at 2×105 cells/well in 96-well plates and incubated for 18 h at 37°C under an atmosphere of 5% CO2 before stimulation. Cells were stimulated with 2 or 10 µg/mL naked siRNA, liposomal siRNA, poly(I : C), or CL097. As negative controls, cells were also incubated with 56.8 or 284 µM empty liposomes in which the cationic core was encapsulated without siRNA. Cells were incubated for another 24 h at 37°C under an atmosphere of 5% CO2. Then, culture supernatants were harvested, and the concentrations of TNF-α and IL-6 were determined by using dedicated enzyme-linked immunosorbent assay (ELISA) kits (eBioscience, San Diego, CA, U.S.A.). For the TLR inhibition assay, CD14+ monocytes were pre-incubated with CU-CPT4a (50 µM), ODN2087 (5 µM), or ODN20958 (5 µM) for 30 min before liposomal siRNA stimulation. These TLR inhibitors can be internalized into the cell without transfection reagents.35–38) Thereafter, cells were incubated for 24 h with 10 µg/mL of liposomal siRNA, poly(I : C), or CL097 along with the above inhibitors. The amount of TNF-α produced was measured by means of an ELISA.
Flow Cytometric AnalysisFor analysis of cell-surface CD14 expression, cells were preincubated with Fc Block and then stained with fluorescein isothiocyanate-conjugated anti-CD14 antibody in fluorescent activated cell sorting (FACS) buffer (phosphate-buffered saline containing 5% FBS and 0.1% sodium azide) for 30 min on ice. After washing twice, the cells were resuspended in 300 µL of FACS buffer containing 7-aminoactinomycin D (0.25 µg/sample), and the fluorescence associated with the live cells was measured on a FACS Calibur system (BD Biosciences). Dead cells and cellular debris were identified by forward scatter/side scatter profiles and 7-aminoactinomycin D staining and were excluded from the analysis. For intracellular staining of TNF-α, cells were stimulated with siRNA, empty liposome, liposomal siRNA, poly(I : C), or CL097 in the presence of the cytokine secretion blocking agent brefeldin A for 6 h. Harvested cells were stained for surface expression of CD14 and then fixed and permeabilized with Cytofix/Cytoperm. Samples were washed twice, followed by staining with phycoerythrin-conjugated anti-TNF-α antibody for 30 min on ice. Cells incubated with an isotype control were used as reference controls. Flow cytometry data were analyzed by using the FlowJo software (FlowJo LLC, Ashland, OR, U.S.A.).
RESULTS AND DISCUSSION
Physicochemical Characteristics, Stability, and Cellular Uptake of the Liposomal siRNACationic core/siRNA complexes (lipoplex siRNA) have been commonly used in vitro gene knockdown studies.39) However, in the case of systemic administration, electrostatic interactions between positively charged lipoplex and negatively charged erythrocytes and proteins in the blood can cause agglutination, inducing embolization.40–42) In this study, therefore, we constructed lipoplex siRNA enveloped in a neutral lipid bilayer of egg phosphatidylcholine and PEG-DSPE, as previously developed by Yagi et al.33) The physicochemical characteristics of the prepared liposomal siRNA are shown in Table 1. The average particle size of the liposomal siRNA was 88.7±1.3 nm and the polydispersity index was less than 0.2, indicating a narrow size distribution. The zeta potential of the liposomal siRNA showed that it was more negatively charged than was the cationic core alone, indicating that the siRNA/cationic core complex was enveloped by a neutral lipid bilayer. siRNA encapsulation efficiency was above 70%.
Table 1. Physicochemical Properties of the Constructed Liposomes
| Particle size (nm) | Polydispersity index | Zeta-potential (mV) |
|---|
| Cationic core | 79.6±0.7 | 0.209±0.007 | 61.2±0.25 |
| Liposomal siRNA | 88.7±1.3 | 0.156±0.010 | −1.06±0.57 |
Each value represents the mean±S.D. (n=3).
Stabilization of siRNA against nuclease degradation is a prerequisite for the use of liposomal siRNA in vivo as a therapeutic application. Therefore, to evaluate the stability of the liposomal siRNA, we incubated it with RNase A, which is present in high concentrations in the blood (Fig. 1A). Naked siRNA and the lipoplex siRNA were digested by RNase A within 30 min; however, siRNA encapsulated within the liposomal carrier remained intact and in its double-stranded form for 24 h. These data confirm that encapsulating siRNA within liposomes may be a useful means of improving the stability of siRNA in the blood circulation.
The cellular uptake of naked A647-siRNA and liposomal A647-siRNA by PBMCs was assessed by using flow cytometry. Within monocytes, the mean fluorescence intensity of liposomal A647-siRNA was significantly higher than that of naked A647-siRNA, indicating that liposomal siRNA, not naked siRNA, is preferentially taken up by monocytes (Fig. 1B). Moreover, a greater uptake of the liposomal siRNA by monocytes than by lymphocytes was observed. The cellular uptake of liposomal siRNA was also confirmed by using the confocal microscopy and by measuring the RNA interference activity of liposomal siRNA (Supplementary Figs. 1, 2). Similar cell type-specific uptake of nanoparticles by PBMCs has been reported by other researchers; for example, Greulich et al. reported that silver nanoparticles (size approx. 70 nm) were detected within monocytes (CD14+) but not within T cells (CD3+), as assessed by light microscopy, flow cytometry, and combined focused ion beam/scanning electron microscopy.27)
PBMC Cytokine Release AssayThe siRNA (β-gal 728) used in the present study contains an immunostimulatory 5′-UGU-3′ motif.13) To examine the innate immune response of human PBMCs to this siRNA, we incubated PBMCs with siRNA, empty liposome, or liposomal siRNA, or poly(I : C) or CL097 as positive controls, and then analyzed the inflammatory cytokine production by ELISA. Liposomal siRNA dose-dependently induced the expression of TNF-α (Fig. 2A) and IL-6 (Fig. 2B), but not interferon-α (data not shown), whereas naked siRNA and empty liposome did not induce any of the cytokines. We also confirmed that lipoplex siRNA induced TNF-α and IL-6 production as well as liposomal siRNA (Supplementary Fig. 3). These results indicate that the induction of cytokine expression was a result of the combination of the siRNA and liposomal carrier, suggesting that internalization of siRNA via a liposomal carrier is an important factor in inducing cytokine production. On the other hand, previous studies have shown that siRNAs have the potential to induce interferon-α production in adherent PBMCs.13,14,16) Therefore, the differences in the cytokine production profile between our study and those of the previous studies are likely the result of differences in the properties of the carrier used. In fact, Judge et al. reported that an siRNA/polyethylenimine complex induced a strong interferon response, whereas an siRNA/poly-L-lysine complex induced a strong TNF-α and IL-6 response13); however, the carrier properties that dictate the siRNA-induced cytokine induction pattern remain unclear. Interestingly, we also observed considerable lot-to-lot variability in cytokine production (Fig. 2). For example, after stimulation with CL097, the amount of TNF-α produced by PBMCs from Lot A was three times that produced by PBMCs from Lot B. A similar trend was seen in IL-6 production (Lot A>C>B), clearly indicating the need for an improved assay to minimize the effects of inter-lot differences in cytokine induction.
Flow Cytometric AnalysisTo identify the causes of the inter-lot differences in cytokine induction, the expression of CD14 (a classical monocyte marker) on the surface of the PBMCs in the three different lots was examined (Figs. 3A, B). CD14 expression was examined because siRNA uptake was much higher in monocytes than in lymphocytes (Fig. 1B). The proportion of CD14+ cells in the total PBMC population decreased in the following order: Lot A>C>B (Fig. 3B), and there was a strong correlation (R2>0.9) between the amount of cytokine produced and the percentage of CD14+ cells in the total PBMC population (Fig. 3C). From these results, we hypothesized that our liposomal siRNA was predominantly taken up by CD14+ monocytes, in which it induced cytokine production, especially the production of TNF-α.
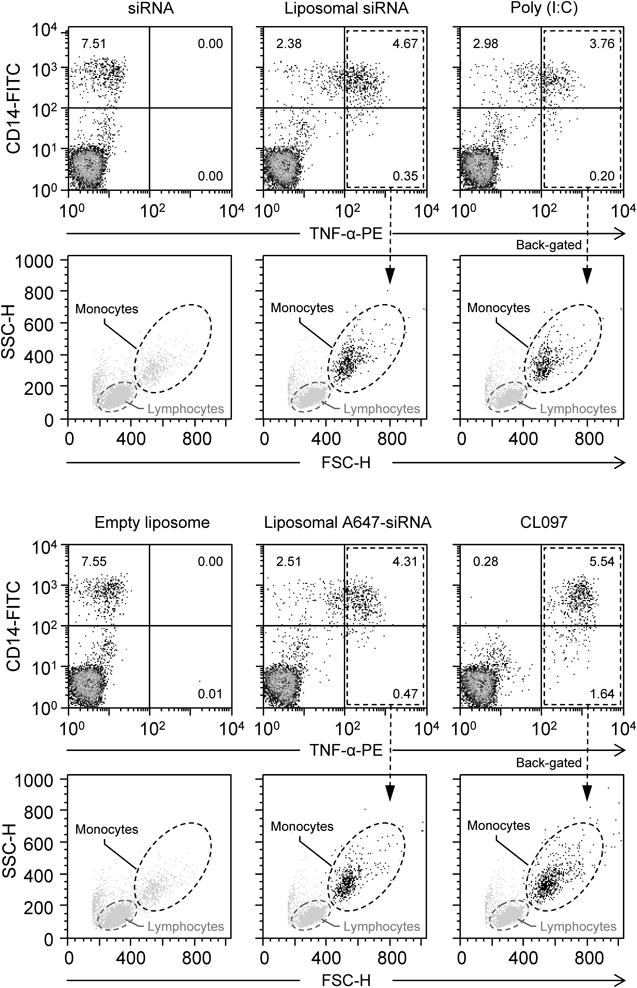
To verify this hypothesis, we identified the TNF-α-producing cells by performing intracellular staining in the presence of brefeldin A, which inhibits protein transport from the endoplasmic reticulum to the Golgi apparatus. No TNF-α-producing cells were detected in PBMCs treated with naked siRNA (Fig. 4), as expected given the low stability (Fig. 1A) and membrane permeability (Fig. 1B) of the naked siRNA. In liposomal siRNA-treated PBMCs, TNF-α-producing cells (gated in the upper- and lower-right quadrants) accounted for approximately 5% of the total PBMC population; more than 90% of TNF-α-producing cells were also positive for CD14 expression (upper-right quadrant). Back-gating of the TNF-α+ population (black dots) onto the original forward scatter/side scatter dot plot (light gray dots) revealed that the majority of the TNF-α-producing cells were monocytes. Additionally, PBMCs treated with liposomal A647-siRNA showed a similar proportion of CD14+ TNF+ double-positive cells, indicating that labeling of the siRNA with Alexa647 did not affect the immune stimulatory activity of the liposomal siRNA. Consistent with the results of the cytokine release assay (Fig. 2A), a TLR3 agonist (poly(I : C)) and a TLR7/8 agonist (CL097) both induced TNF-α production in the CD14+ monocytes (Fig. 4). Together, these results demonstrate that CD14+ monocytes are the major producers of TNF-α after induction by our liposomal siRNA.
CD14 is a glycosylphosphatidylinositol-anchored membrane-associated protein that promotes the uptake of nucleic acids and acts as a co-receptor for endosomal TLR activation.43,44) Human CD14+ monocytes are part of the mononuclear phagocyte system; they specialize in phagocytosis, the production of reactive oxygen species, and the secretion of inflammatory cytokines in response to a broad range of microbial cues.45) Recently, Cros et al. characterized the CD14+ and CD14dimCD16+ monocyte subpopulations as “inflammatory” and “patrolling” monocytes, respectively.45) As shown in Fig. 4, in the CL097-treated PBMCs, the majority of the TNF-α-producing cells were in the CD14+ population (upper-right quadrant; 5.54%), with some in the CD14dim population (lower-right quadrant; 1.64%). Although further examination is needed, these results indicate that not only CD14+ inflammatory monocytes but also CD14dim patrolling monocytes produced TNF-α via TLR7 and TLR8 in response to CL097; however, in comparison, fewer CD14dim monocytes responded to our liposomal siRNA (0.35%, Fig. 4).
Cytokine Release Assay Using CD14+ MonocytesSince high inter-lot variability in PBMC cytokine release is a major issue for the cytokine release assay used in this study, we examined whether the use of CD14+ monocytes purified from the total PBMC population, could reduce the inter-lot variability. Figure 5A shows the TNF-α production by commercially available CD14+ monocytes from three individual lots; in accordance with the results of the TNF-α production by PBMC assay, TNF-α was secreted by CD14+ monocytes stimulated with liposomal siRNA, poly(I : C), or CL097. The results show that the inter-lot variability in TNF-α production was markedly lower when CD14+ monocytes were used than when the total PBMC population was used (Fig. 5A). Although the use of pooled PBMCs from different donors might mitigate inter-assay variability, our results suggest that CD14+ monocytes will be more useful for characterizing the immunostimulatory properties of siRNA because of their inherently lower inter-lot variability. However, this result does not eliminate the rationale of using PBMCs, because, compared with CD14+ monocytes, PBMCs more accurately reflect actual physiological conditions and therefore are useful for determining cytokine production profiles that can then be used to predict which adverse effects are likely to occur in vivo.
To identify the siRNA recognition receptors on CD14+ monocytes, we examined the inhibitory effects of TLR antagonists on the TNF-α release induced by the liposomal siRNA and found that there was no marked decrease in cell viability (cell viability, >95%) when the CD14+ monocytes were incubated with the TLR antagonists (data not shown). Liposomal siRNA-induced TNF-α production was inhibited by ODN2087 (TLR7/TLR8 antagonists), but not by ODN20958 (TLR7 antagonist) or CU-CPT4a (TLR3 antagonist) (Fig. 5B); thus in CD14+ monocytes, immunostimulatory siRNA encapsulated in liposome was dominantly recognized by endosomal TLR8. These results are consistent with a previous quantitative analysis of mRNA expression that demonstrated that monocytes were characterized by high expression of TLR8 but very low expression of TLR3 and TLR7.46)
Primary monocyte and PBMC cultures have a limited number of passages. To overcome this limitation, cell lines are often used as models of primary cells. However, this results in differences developing between the primary and immortalized cell lines. For example, peripheral blood monocytes produce a greater amount of inflammation-related cytokines than do THP-1 monocytes.47) Moreover, Judge et al. failed to detect signaling by β-gal 728 siRNA duplexes in stable HEK293 cells overexpressing TLR3, TLR7, or TLR8, even though a signal was detected with positive controls (TLR agonistic ligands).13) Why the TLRs were unable to recognize siRNA in the transfected cell line remains unclear; however, a recent structural and biological study of TLR8 by Tanji et al. suggests that TLR8 recognizes a short oligonucleotide at two distinct sites, both of which are essential for single-stranded RNA recognition, but only one of which is essential for the recognition of chemical ligands.48) In addition, it has also been reported that endosomal localization and proteolytic cleavage of TLR8 are indispensable for nucleic acid sensing and signaling initiation.49,50) In HEK293 cells transfected with TLR8, the cleavage products of TLR8 are reportedly undetectable, and there is no activation of TLR8 in response to single-stranded RNA.49) Therefore, the use of a primary cell culture is currently thought to be the best way to evaluate immune response to siRNA in vitro. Hopefully, further elucidation of these mechanisms will lead to a better understanding of TLR signaling and the development of a standardized, simple, and accurate assay for immune response to liposomal siRNA.
In conclusion, we identified CD14+ monocytes as the major producer of TNF-α induced by a liposome-encapsulated immunostimulatory siRNA. Our results suggest that the peripheral blood CD14+ monocyte-based cytokine release assay is a specific means of alleviating the lot-to-lot variability in the cytokine release profiles of commercially available PBMCs. Our results also show that the peripheral blood CD14+ monocyte-based cytokine release assay can be used to identify the siRNA recognition receptors that mediate individual cytokine production.
Acknowledgments
This work was funded in part with Research Grants for the Research on Regulatory Harmonization and Evaluation of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetics from Japan Agency for Medical Research and Development, AMED, and Initiative for Accelerating Regulatory Science in Innovative Drug, Medical Device, and Regenerative Medicine from the Japan Ministry of Health, Labour and Welfare. We thank Prof. T. Yamaguchi for his valuable advice on our work.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498 (2001).
- 2) Takeshita F, Ochiya T. Therapeutic potential of RNA interference against cancer. Cancer Sci., 97, 689–696 (2006).
- 3) Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature, 457, 426–433 (2009).
- 4) Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev., 87, 108–119 (2015).
- 5) Kowalski PS, Leus NG, Scherphof GL, Ruiters MH, Kamps JA, Molema G. Targeted siRNA delivery to diseased microvascular endothelial cells: cellular and molecular concepts. IUBMB Life, 63, 648–658 (2011).
- 6) Raemdonck K, Vandenbroucke RE, Demeester J, Sanders NN, De Smedt SC. Maintaining the silence: reflections on long-term RNAi. Drug Discov. Today, 13, 917–931 (2008).
- 7) Zhou J, Shum KT, Burnett JC, Rossi JJ. Nanoparticle-based delivery of RNAi therapeutics: Progress and challenges. Pharmaceuticals (Basel), 6, 85–107 (2013).
- 8) Chiu YL, Rana TM. siRNA function in RNAi: A chemical modification analysis. RNA, 9, 1034–1048 (2003).
- 9) Cho WG, Albuquerque RJ, Kleinman ME, Tarallo V, Greco A, Nozaki M, Green MG, Baffi JZ, Ambati BK, De Falco M, Alexander JS, Brunetti A, De Falco S, Ambati J. Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. Proc. Natl. Acad. Sci. U.S.A., 106, 7137–7142 (2009).
- 10) Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov., 9, 57–67 (2010).
- 11) Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature, 452, 591–597 (2008).
- 12) Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol., 9, 535–542 (2009).
- 13) Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol., 23, 457–462 (2005).
- 14) Sioud M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J. Mol. Biol., 348, 1079–1090 (2005).
- 15) Karikó K, Bhuyan P, Capodici J, Weissman D. Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through Toll-like receptor 3. J. Immunol., 172, 6545–6549 (2004).
- 16) Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med., 11, 263–270 (2005).
- 17) Forsbach A, Nemorin JG, Montino C, Muller C, Samulowitz U, Vicari AP, Jurk M, Mutwiri GK, Krieg AM, Lipford GB, Vollmer J. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol., 180, 3729–3738 (2008).
- 18) Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science, 303, 1526–1529 (2004).
- 19) Govindaraj RG, Manavalan B, Basith S, Choi S. Comparative analysis of species-specific ligand recognition in Toll-like receptor 8 signaling: a hypothesis. PLoS ONE, 6, e25118 (2011).
- 20) Zhu J, Brownlie R, Liu Q, Babiuk LA, Potter A, Mutwiri GK. Characterization of bovine Toll-like receptor 8: ligand specificity, signaling essential sites and dimerization. Mol. Immunol., 46, 978–990 (2009).
- 21) Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, Bauer S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol., 3, 499 (2002).
- 22) Goodchild A, Nopper N, King A, Doan T, Tanudji M, Arndt GM, Poidinger M, Rivory LP, Passioura T. Sequence determinants of innate immune activation by short interfering RNAs. BMC Immunol., 10, 40 (2009).
- 23) Judge AD, Bola G, Lee AC, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther., 13, 494–505 (2006).
- 24) Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity, 23, 165–175 (2005).
- 25) Sakurai Y, Hatakeyama H, Sato Y, Hyodo M, Akita H, Harashima H. Gene silencing via RNAi and siRNA quantification in tumor tissue using MEND, a liposomal siRNA delivery system. Mol. Ther., 21, 1195–1203 (2013).
- 26) Dobrovolskaia MA, McNeil SE. Strategy for selecting nanotechnology carriers to overcome immunological and hematological toxicities challenging clinical translation of nucleic acid-based therapeutics. Expert Opin. Drug Deliv., 12, 1163–1175 (2015).
- 27) Greulich C, Diendorf J, Geßmann J, Simon T, Habijan T, Eggeler G, Schildhauer TA, Epple M, Köller M. Cell type-specific responses of peripheral blood mononuclear cells to silver nanoparticles. Acta Biomater., 7, 3505–3514 (2011).
- 28) Xue HY, Liu S, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine (Lond.), 9, 295–312 (2014).
- 29) Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I, Fox B, Tarrant G, Robinson J, Meager T, Dolman C, Thorpe SJ, Bristow A, Wadhwa M, Thorpe R, Poole S. “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J. Immunol., 179, 3325–3331 (2007).
- 30) Dobrovolskaia MA, McNeil SE. Understanding the correlation between in vitro and in vivo immunotoxicity tests for nanomedicines. J. Control. Release, 172, 456–466 (2013).
- 31) Schmal H, Niemeyer P, Roesslein M, Hartl D, Loop T, Sudkamp NP, Stark GB, Mehlhorn AT. Comparison of cellular functionality of human mesenchymal stromal cells and PBMC. Cytotherapy, 9, 69–79 (2007).
- 32) Jeffs LB, Palmer LR, Ambegia EG, Giesbrecht C, Ewanick S, MacLachlan I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm. Res., 22, 362–372 (2005).
- 33) Yagi N, Manabe I, Tottori T, Ishihara A, Ogata F, Kim JH, Nishimura S, Fujiu K, Oishi Y, Itaka K, Kato Y, Yamauchi M, Nagai R. A nanoparticle system specifically designed to deliver short interfering RNA inhibits tumor growth in vivo. Cancer Res., 69, 6531–6538 (2009).
- 34) Yamauchi M, Kusano H, Saito E, Iwata T, Nakakura M, Kato Y, Uochi T, Akinaga S, Aoki N. Improved formulations of antisense oligodeoxynucleotides using wrapped liposomes. J. Control. Release, 114, 268–275 (2006).
- 35) Jurk M, Kritzler A, Schulte B, Tluk S, Schetter C, Krieg AM, Vollmer J. Modulating responsiveness of human TLR7 and 8 to small molecule ligands with T-rich phosphorothiate oligodeoxynucleotides. Eur. J. Immunol., 36, 1815–1826 (2006).
- 36) Juliano RL, Ming X, Nakagawa O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug. Chem., 23, 147–157 (2012).
- 37) Hackstein H, Knoche A, Nockher A, Poeling J, Kubin T, Jurk M, Vollmer J, Bein G. The TLR7/8 ligand resiquimod targets monocyte-derived dendritic cell differentiation via TLR8 and augments functional dendritic cell generation. Cell. Immunol., 271, 401–412 (2011).
- 38) Cheng K, Wang X, Yin H. Small-molecule inhibitors of the TLR3/dsRNA complex. J. Am. Chem. Soc., 133, 3764–3767 (2011).
- 39) Schroeder A, Levins CG, Cortez C, Langer R, Anderson DG. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med., 267, 9–21 (2010).
- 40) Ogris M, Wagner E. Targeting tumors with non-viral gene delivery systems. Drug Discov. Today, 7, 479–485 (2002).
- 41) Litzinger DC, Brown JM, Wala I, Kaufman SA, Van GY, Farrell CL, Collins D. Fate of cationic liposomes and their complex with oligonucleotide in vivo. Biochim. Biophys. Acta, 1281, 139–149 (1996).
- 42) Hatanaka K, Asai T, Koide H, Kenjo E, Tsuzuku T, Harada N, Tsukada H, Oku N. Development of double-stranded siRNA labeling method using positron emitter and its in vivo trafficking analyzed by positron emission tomography. Bioconjug. Chem., 21, 756–763 (2010).
- 43) Baumann CL, Aspalter IM, Sharif O, Pichlmair A, Bluml S, Grebien F, Bruckner M, Pasierbek P, Aumayr K, Planyavsky M, Bennett KL, Colinge J, Knapp S, Superti-Furga G. CD14 is a coreceptor of Toll-like receptors 7 and 9. J. Exp. Med., 207, 2689–2701 (2010).
- 44) Lee HK, Dunzendorfer S, Soldau K, Tobias PS. Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity, 24, 153–163 (2006).
- 45) Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, Jais JP, D’Cruz D, Casanova JL, Trouillet C, Geissmann F. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity, 33, 375–386 (2010).
- 46) Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of Toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol., 168, 4531–4537 (2002).
- 47) Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm., 2013, 697972 (2013).
- 48) Tanji H, Ohto U, Shibata T, Taoka M, Yamauchi Y, Isobe T, Miyake K, Shimizu T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol., 22, 109–115 (2015).
- 49) Ishii N, Funami K, Tatematsu M, Seya T, Matsumoto M. Endosomal localization of TLR8 confers distinctive proteolytic processing on human myeloid cells. J. Immunol., 193, 5118–5128 (2014).
- 50) Itoh H, Tatematsu M, Watanabe A, Iwano K, Funami K, Seya T, Matsumoto M. UNC93B1 physically associates with human TLR8 and regulates TLR8-mediated signaling. PLoS ONE, 6, e28500 (2011).