Abstract
We recently demonstrated that activation of spinal sigma-1 receptors (Sig-1Rs) induces pain hypersensitivity via the activation of neuronal nitric oxide synthase (nNOS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (Nox2). However, the potential direct interaction between nNOS-derived nitric oxide (NO) and Nox2-derived reactive oxygen species (ROS) is poorly understood, particularly with respect to the potentiation of N-methyl-D-aspartate (NMDA) receptor activity in the spinal cord associated with the development of central sensitization. Thus, the main purpose of this study was to investigate whether Sig-1R-induced and nNOS-derived NO modulates spinal Nox2 activation leading to an increase in ROS production and ultimately to the potentiation of NMDA receptor activity and pain hypersensitivity. Intrathecal pretreatment with the nNOS inhibitor, 7-nitroindazole or with the Nox inhibitor, apocynin significantly inhibited the mechanical and thermal hypersensitivity induced by intrathecal administration of the Sig-1R agonist, 2-(4-morpholinethyl) 1-phenylcyclohexanecarboxylate hydrochloride (PRE084). Conversely, pretreatment with 5,10,15,20-tetrakis-(4-sulphonatophenyl)-porphyrinato iron(III) (FeTPPS; a scavenger of peroxynitrite, a toxic reaction product of NO and superoxide) had no effect on the PRE084-induced pain hypersensitivity. Pretreatment with 7-nitroindazole significantly reduced the PRE084-induced increase in Nox2 activity and concomitant ROS production in the lumbar spinal cord dorsal horn, whereas apocynin did not alter the PRE084-induced changes in nNOS phosphorylation. On the other hand pretreatment with apocynin suppressed the PRE084-induced increase in the protein kinase C (PKC)-dependent phosphorylation of NMDA receptor GluN1 subunit (pGluN1) at Ser896 site in the dorsal horn. These findings demonstrate that spinal Sig-1R-induced pain hypersensitivity is mediated by nNOS activation, which leads to an increase in Nox2 activity ultimately resulting in a ROS-induced increase in PKC-dependent pGluN1 expression.
Nitric oxide (NO) is involved in many pathophysiological processes, including the important role that it serves as a neurotransmitter/neuromodulator involved in nociceptive processes in the spinal cord dorsal horn.1) It has been well demonstrated that NO is a diffusible gas synthesized in biological systems by enzymes known as nitric oxide synthases (NOS) (neuronal/nNOS, inducible/iNOS, or endothelial/eNOS).1) Interestingly, nNOS co-localizes with N-methyl-D-aspartic acid (NMDA) receptors and is functionally coupled to this glutamate receptor subtype via binding with postsynaptic density protein-95.2) Several studies suggest that activation of NMDA receptors modulates spinal nociceptive transmission via nNOS, which is dephosphorylated and activated by the Ca2+/calmodulin (CaM)-dependent phosphatase calcineurin, resulting from Ca2+ influx through NMDA receptors.3–5) In a previous study, we showed that blockade of spinal nNOS using intrathecal (i.t.) administration of the nNOS inhibitor, 7-nitroindazole, reduces sigma-1 receptor (Sig-1R)-induced pain hypersensitivity and suppresses the Sig-1R-induced increase in protein kinase C (PKC)-dependent phosphorylation of GluN1 subunit (pGluN1) resulting in the potentiation of NMDA receptor function.6) These findings raise the possibility that the action of NO on nociceptive transmission at spinal synapses could be closely linked to Sig-1R-induced functional activation of NMDA receptors.
It has been suggested that reactive oxygen species (ROS) play an important role in ‘central sensitization,’ a critical mechanism underlying persistent pain, via increases in NMDA receptor phosphorylation in the spinal cord dorsal horn.7) ROS can be generated by activation of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) enzyme.8) Among six homologs, the NADPH oxidase 2 (Nox2) isoform has been indicated as the main source of ROS in neurons, which produces superoxide following neuronal NMDA receptor activation.9,10) Nox2 is a highly regulated membrane bound enzyme complex that is composed of membrane-bound subunits (p22phox and gp91phox) and cytosolic subunits (p40phox, p47phox, and p67phox) as well as the small guanosine triphosphatase (GTPase) Rac. The cytosolic components translocate to the cell membrane in order to form a functional active enzyme complex.11,12) In particular p47phox has an important role in both translocation and assembly, by which Nox enzyme activation is initiated.10) In a previous study, we demonstrated that spinal Sig-1R activation increases p47phox translocation to the membrane fraction and concomitant ROS production.13) However, it remains unclear whether Nox2-derived ROS mediates Sig-1R-induced potentiation of NMDA receptor function.
The activation of spinal Sig-1Rs has been considered to play an important role in pain sensitization.6,14) As previously described, Sig-1R-induced pain is closely associated with nNOS activation as well as Nox2-derived ROS production and both ROS and NO participate in NMDA receptor signaling.9) However, the hypothetical interaction between these two signaling pathways is poorly understood, particularly as it relates to the potentiation of NMDA receptor activity in the spinal cord. The main goal of this study was to investigate whether nNOS-derived NO induced by Sig-1R stimulation plays an important role in spinal Nox2 activation leading to increases in ROS production and the potentiation of NMDA receptor activity and ultimately resulting in pain hypersensitivity. In this regard, the present study specifically examined whether: (1) nNOS and Nox activation or peroxynitrite, a reaction product of NO and superoxide, mediates Sig-1R-induced pain hypersensitivity; (2) nNOS-derived NO induced by Sig-1R activation increases Nox2 activity and concomitant ROS production in the spinal cord dorsal horn; and finally, (3) Nox-derived ROS contributes to the increase in phosphorylation of NMDA receptors.
MATERIALS AND METHODS
Experimental AnimalsMale ICR mice (20–25 g) were purchased from the Laboratory Animal Center of Seoul National University (Seoul, Republic of Korea) and used for the present study. Mice had free access to food and water and were kept in temperature and light controlled rooms (23±2°C, 12/12 h light/dark cycle with lights on at 08:00) for at least 3 d prior to the beginning of each experiment. The experimental protocols for animal usage were reviewed and approved by the SNU Animal Care and Use Committee and conform to NIH guidelines (1985).15)
Drugs and Intrathecal (i.t.) AdministrationThe following drugs were used: 2-(4-morpholinethyl)1-phenylcyclohexanecarboxylate (PRE084; PRE, 3 nmol), a Sig-1R agonist; 7-nitroindazole (7-NI, 200 nmol), a specific nNOS inhibitor; L-N6-(1-iminoethyl)lysine dihydrochloride (L-NIL, 20 nmol), a selective iNOS inhibitor; diphenyleneiodonium chloride (DPI, 6 nmol), an eNOS inhibitor; 4′-hydroxy-3′-methoxyacetophenone (apocynin, 100 nmol), a NADPH oxidase inhibitor; 5,10,15,20-tetrakis-(4-sulphonatophenyl)-porphyrinato iron(III) (FeTPPS, 3 nmol), a peroxynitrite scavenger; chelerythrine (CHE, 1 nmol), a PKC inhibitor. Apocynin, 7-NI and L-NIL were supplied by Sigma-Aldrich (St. Louis, MO, U.S.A.) and DPI by MP Biomedicals (OH, U.S.A.). PRE and CHE were obtained from Tocris Cookson Ltd. (Bristol, U.K.) and FeTPPS from Calbiochem (San Diego, CA, U.S.A.). The doses of all drugs were selected based on or slightly modified from established doses previously used in the literature including those used in previous studies from our laboratories.6,13,16–19) PRE, L-NIL and FeTPPS were dissolved in physiological saline. 7-NI was dissolved in 5% dimethyl sulfoxide (DMSO) in Corn oil. DPI, apocynin and CHE were dissolved in 5% DMSO in physiological saline. The control group received an injection of vehicle.
All drugs were dissolved in 5 µL of vehicle and were administered intrathecally 10 min before PRE injection. Intrathecal injections were performed at the L5–6 intervertebral space of unanesthetized mice using a Hamilton syringe connected to a 30-gauge needle as previously described by Hylden and Wilcox.20) Animals were randomly assigned to experimental groups and subsequent drug treatment and analysis were performed blindly.
Mechanical and Thermal Hypersensitivity TestPain hypersensitivity was measured before and at 30, 60 and 120 min after administration of the Sig-1R agonist, PRE (or vehicle) in all groups of mice (n=5 mice/group). Sensitization to innocuous mechanical stimulation was examined using a von Frey filament (North Coast Medical, Morgan Hill, CA, U.S.A.) as previously described.6) Based on this previous study, the 0.16 g von Frey filament was selected for testing and was applied 10 times to each hind paw with each application separated by 10 s intervals. Then, the number of paw withdrawal responses was counted. The results of mechanical response testing in each experimental animal were expressed as a percent paw withdrawal frequency (PWF, %), which represented the percentage of paw withdrawals out of a maximum of 20 (right hind paw-10 and left hind paw-10).
Sensitization to noxious heat stimulation was examined with a hot-plate apparatus (Model-35100, Ugo Basile, Comerio, Italy). The temperature of the hot plate was maintained at 55±0.5°C. Animals were placed into an acrylic cylinder (20 cm in diameter, 25 cm high) on the heated surface, and the time between placement and the initial shaking, licking or lifting of their hind paws or jumping (whichever occurred first), was recorded as the latency responses (s). The test was duplicated in each animal, and the mean latency response was calculated. Cutoff time in the absence of a response was set at 20 s to minimize potential tissue damage.
Western Blot AssayMice were deeply anesthetized with 3% isoflurane 30 min after i.t. injection of PRE084 (n=6 mice/group). The Western blot assay was performed as described in our previous report.13) The spinal cord was extracted by pressure expulsion with air into an ice-cooled, saline-filled glass dish. Following expulsion, the dorsal horns from the L4–6 spinal segments were homogenized in lysis buffer (20 mM Tris–HCl, 10 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 2 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4, and proteinase inhibitors), and homogenates were centrifuged at 15000 rpm for 40 min at 4°C. For preparation of the membrane fraction, the pellet was re-suspended in lysis buffer containing 1% Triton X-100. For preparation of total proteins, the spinal cord dorsal horns were homogenized first in lysis buffer containing 1% Triton X-100. Homogenates were subsequently centrifuged using the same method as described above and the supernatant was used for Western blot analysis.
The total amount of protein in each sample was determined using the Bradford dye assay. Spinal cord homogenates (25–35 µg protein) were separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membrane. After the blots had been washed with TBST (10 mM Tris–HCl, pH 7.6, 150 mM NaCl, and 0.05% Tween-20), the membranes were blocked with 5% skimmed milk for 1 h at room temperature and incubated at 4°C overnight with a primary antibody specific for p47phox (rabbit polyclonal anti-p47phox antibody, 1:1k, cat# 14015, Santa Cruz Biotechnology Inc.), PKC-dependent pGluN1 (rabbit polyclonal anti-pGluN1 Ser896 antibody, 1:1k, cat# ABN88, Millipore Co., U.S.A.) or β-actin (mouse monoclonal anti-β-actin antibody, 1:5k, cat# 47778, Santa Cruz Biotechnology Inc., U.S.A.). After washing with TBST, membranes were incubated for 4 h with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse antibody (1:10k, Santa Cruz Biotechnology Inc.). The bands were visualized with enhanced chemiluminescence (Amersham Biosciences, Buckinghamshire, U.K.). The positive pixel area of specific bands was measured with a computer-assisted image analysis system and normalized against the corresponding β-actin loading control bands. To analyze activation of Nox2, the ratio of the membrane fraction (M) of p47phox to the total protein (T) of p47phox expression was calculated. The value of the ratio of p47phox (M) to p47phox (T) expression in the control groups was set at 100%. Thus, the percent change in p47phox (M) to p47phox (T) expression was examined for each group. For analysis of pGluN1 expression, the value of the control groups was set at 100% and, then, the percent change relative to the control groups was calculated for each group.
ImmunohistochemistryThirty minutes after i.t. injection of PRE084 mice were anesthetized with 3% isoflurane and spinal cord immunohistochemistry was performed as described in our previous report (n=5–7 mice/group).6) Briefly mice were perfused transcardially with calcium-free Tyrode’s solution followed by 200 mL of a fixative containing 4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer (pH 7.4). The spinal cords were removed immediately after perfusion, post-fixed in the identical fixative overnight and then placed in 30% sucrose in phosphate buffered saline (PBS) (pH 7.4) for 24 h at 4°C. Serial transverse sections (40 µm) of the spinal cord were cut using a cryostat (Microm, Germany). L4–6 spinal tissue sections were incubated at 4°C with a primary antibody specific for phosphorylated form of nNOS (pnNOS) (rabbit polyclonal anti-pnNOS Ser847 antibody, 1:3k, cat# ab16650, Abcam Inc.) or PKC-dependent pGluN1 (rabbit polyclonal anti-pGluN1 Ser896 antibody, 1:1k, cat# 06–640, Upstate Biotechnology). The primary antibodies were detected by incubating the tissue in biotinylated anti-rabbit immunoglobulin G (IgG) (1 : 200, cat# BA-1000; Vector Laboratories, Inc.) for 1 h at r.t. Then following several rinses, the tissue was further incubated in avidin–biotin–peroxidase complex (ABC) for 1 h. Visualization of the ABC complex was performed using 3,3-diaminobenzidine (Sigma) and the 3,3-diaminobenzidine reaction was subsequently intensified with 0.2% nickel chloride.
Detection of in Situ Generation of ROSTo detect the in situ generation of ROS in the spinal cord, dihydroethidium (DHE) staining was performed as previously described (n=4 mice/group).13) Thirty minutes after i.t. injection of PRE084 mice were anesthetized with 3% isoflurane and perfused transcardially with calcium-free Tyrode’s solution. The lumbar spinal cord from each mouse was collected and embedded in Tissue-Tek O.C.T. compound (Sakura Finetechnical Co., Japan) and then frozen immediately in liquid nitrogen. Spinal cord segments were cut into 40 µm thick transverse sections and placed on a glass slide. DHE (1 nM, cat# D-1168, Invitrogen) was topically applied to each section and cover-slipped. Slides were incubated in a dark, humidified chamber at 37°C for 30 min. DHE is oxidized by superoxide to ethidium bromide, which binds to the DNA and emits red fluorescence.
Image AnalysisTissue sections were first examined under darkfield microscopy at 4× magnification to determine the gray matter landmarks and to distinguish the laminae of the dorsal horn as defined by Abbadie and Besson.21) The immunostaining of pnNOS and pGluN1 in the spinal cord was then examined under a brightfield microscope. The red fluorescence associated with DHE staining was detected through a 580 nm long-pass filter and examined using a fluorescence microscope (Zeiss Axioscope, Hallbergmoos, Germany). Three to five spinal cord sections from the L4–6 spinal cord segments were randomly selected from each animal, and subsequently scanned using a cooled CCD camera (Micromax Kodak 1317; Princeton Instruments, AZ, U.S.A.) connected to a computer-assisted image analysis system (Metamorph version 6.3r2; Molecular Devices Corporation, PA, U.S.A.). To maintain a constant threshold for each immunostained image and to compensate for subtle variability of the immunostaining, we only counted cells that were at least 70% darker than the average gray level of each image after background subtraction and shading correction. The average number of pnNOS-immunoreactive (ir) or pGluN1-ir cells and the average intensity of DHE fluorescence per section from each animal were obtained in the following three spinal cord dorsal horn regions (as defined by Abbadie and Besson21)): 1) the superficial dorsal horn (SDH, laminae I–II); 2) the nucleus proprius (NP, laminae III–IV); and 3) the neck region (NECK, laminae V–VI). The values were averaged across each group and presented as group data. All analytical procedures described above and the image analysis of individual sections were performed by an investigator who was blinded to the experimental conditions.
Measurement of NO in the Spinal CordNO was determined in the spinal cord dorsal horns from the lumbar enlargement using a nitric oxide detection kit (cat# ADI-917-020, Enzo Life Sciences Inc., U.S.A.). This nitric oxide (total), detection kit is based on the enzymatic conversion of nitrate to nitrite by the enzyme nitrate reductase, followed by the Griess reaction to form a colored azo dye product. Animals (n=4–6 mice/group) were euthanized 30 min after PRE084 injection. Mice were deeply anesthetized with 3% isoflurane in a mixture of N2O/O2 gas and perfused transcardially with calcium-free Tyrode’s solution. The isolated spinal cords were homogenized in PBS by sonication. Homogenates were subsequently centrifuged at 400×g for 10 min at 4°C and, then, the supernatant was used for nitric oxide detection following the manufacturer’s recommendation.
Statistical AnalysisData are expressed as the mean±standard error of the mean (S.E.M.) Statistical analyses were performed by using Prism 5.0 (Graph Pad Software, San Diego, U.S.A.). Repeated measures two-way ANOVA was performed to determine differences in the data from the time course changes in pain hypersensitivity shown in Fig. 2. Statistical analyses for other data were performed with one-way ANOVA. Post-hoc analysis was performed using the Bonferroni’s multiple comparison test in order to determine the p-value among experimental groups. ANOVA values and degrees of freedom were shown in Supplementary materials Table S1 and S2. A p<0.05 was considered statistically significant.
RESULTS
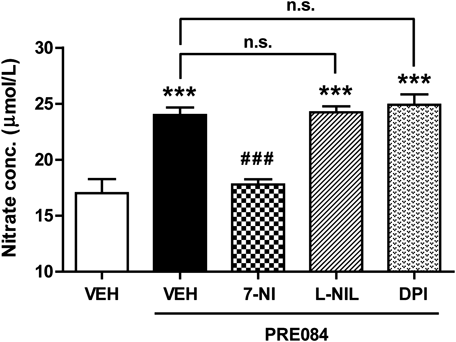
Sig-1R Activation Increases NO Production in the Spinal Cord Dorsal Horn through nNOS ActivationTo investigate whether spinal Sig-1R activation modulates NO production in the spinal cord, we examined the effect of i.t. administration of the Sig-1R agonist, PRE084 on NO levels in the dorsal horn by examining changes in nitrate concentration. Intrathecal administration of PRE084 (3 nmol) significantly increased NO production in the dorsal horn 30 min after PRE084 injection as compared with the vehicle-treated group (Fig. 1; *** p<0.001 vs. VEH+VEH). NO is synthesized in biological systems by three NOS enzymes (nNOS, iNOS, or eNOS).1) In order to assess which of these isoforms is most relevant to this Sig-1R-induced increase in NO production, we next examined the effects of i.t. pretreatment with an nNOS, iNOS and eNOS inhibitor on the Sig-1R-induced increase in NO production in the dorsal horn. As illustrated in Fig. 1, i.t. pretreatment with the nNOS inhibitor, 7-NI (200 nmol) significantly reduced this PRE084-induced increase in NO production (Fig. 1; ### p<0.001 vs. VEH+PRE). On the other hand, pretreatment with the iNOS inhibitor, L-NIL (20 nmol) or the eNOS inhibitor, DPI (6 nmol) had no effect on the Sig-1R agonist-induced increase in spinal NO production (Fig. 1; *** p<0.001 vs. VEH+VEH; non-significance vs. VEH+PRE). Moreover, the i.t. injection of these inhibitors alone, in the absence of PRE, did not affect NO production in comparison to the VEH+VEH group (data not shown). These results demonstrate that the Sig-1R-induced increase in NO production is most closely associated with nNOS activation in the spinal cord dorsal horn.
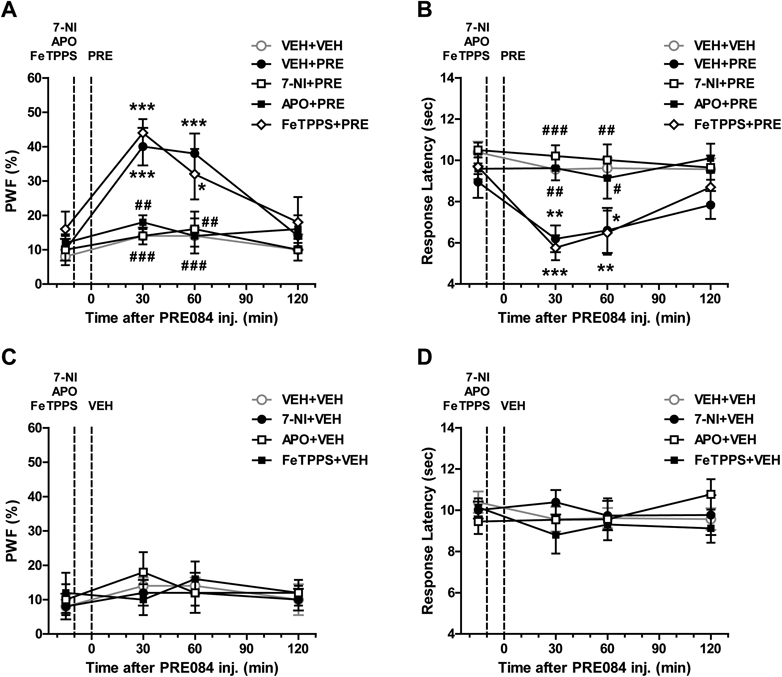
Sig-1R Activation Induces Nociceptive Hypersensitivity through nNOS and Nox Activation, but Peroxynitrite Does Not Contribute to This Increased Pain SensitivityIntrathecal administration of the Sig-1R agonist, PRE084 (3 nmol) significantly increased paw withdrawal frequency (PWF, %) to innocuous mechanical stimuli (Fig. 2A) and decreased response latency (s) to noxious heat stimuli (Fig. 2B) at 30 and 60 min after PRE084 injection as compared with those of the vehicle-treated group (Figs. 2A, B; * p<0.05, ** p<0.01, *** p<0.001 vs. VEH+VEH). Pretreatment with the nNOS inhibitor, 7-NI (200 nmol) or the Nox inhibitor, apocynin (APO, 100 nmol) significantly reduced this PRE084-induced pain hypersensitivity (Figs. 2A, B; # p<0.05, ## p<0.01, ### p<0.001 vs. VEH+PRE). To further investigate whether the peroxynitrite, which is synthesized by the interaction of NO and superoxide, contributes to this pain hypersensitivity, we next examined the effect of i.t. pretreatment with a peroxynitrite scavenger on the PRE084-induced pain hypersensitivity. As shown in Figs. 2A and B, i.t. pretreatment with the peroxynitrite scavenger, FeTPPS (3 nmol) had no effect on the PRE084-induced pain hypersensitivity as compared with that of the PRE084-treated group that received vehicle (Figs. 2A, B; * p<0.05, ** p<0.01, *** p<0.001 vs. VEH+VEH; non-significance vs. VEH+PRE). The i.t. injection of these inhibitors alone (7-NI+VEH, APO+VEH, FeTPPS+VEH), in the absence of PRE, did not affect pain hypersensitivity in comparison to the VEH+VEH group (Figs. 2C, D).
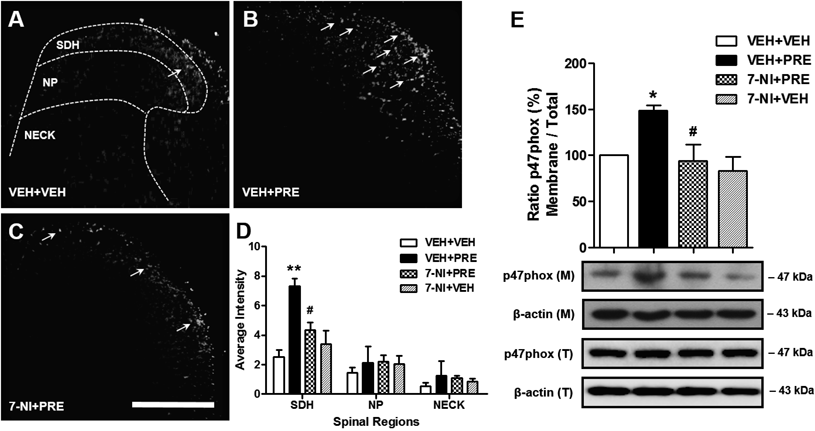
Sig-1R-Induced nNOS Activation Increases ROS Production and Nox2 Activity in the Spinal Cord Dorsal HornTo investigate whether Sig-1R-induced NO modulates ROS production, we examined the effects of i.t. pretreatment with an nNOS inhibitor on the Sig-1R-induced increase in ROS production in the dorsal horn by measuring the fluorescence of DHE staining. As illustrated in Fig. 3, i.t. pretreatment with the nNOS inhibitor, 7-NI (200 nmol, 7-NI+PRE) significantly inhibited the PRE084-induced increase in ROS production in the SDH (laminae I–II) region of the lumbar spinal cord dorsal horn (Figs. 3A–D; ** p<0.01 vs. VEH+VEH, # p<0.05 vs. VEH+PRE). The i.t. injection of this inhibitor alone (7-NI+VEH), in the absence of PRE, did not affect ROS production in comparison to the VEH+VEH group.
To investigate whether the NO induced by Sig-1R stimulation modulates Nox2 activation, we next examined the effects of i.t. pretreatment with 7-NI on the Sig-1R-induced increase in Nox2 activation in the dorsal horn by measuring the expression of the Nox2 subunit p47phox using a Western blot assay. Since an increase in the membrane fraction of the p47phox represents an increase in the active forms of the Nox2 enzyme and since the total protein expression of p47phox did not change following PRE084 injection, then the increase in the ratio of membrane (M) to total (T) protein expression of p47phox is considered to reflect an increase in the active forms of Nox2. As illustrated in Fig. 3E, i.t. pretreatment with 7-NI (200 nmol, 7-NI+PRE) significantly inhibited the PRE084-induced increase in the ratio of membrane to total protein expression of p47phox in the lumbar spinal cord dorsal horn (Fig. 3E; * p<0.05 vs. VEH+VEH, # p<0.05 vs. VEH+PRE). The i.t. injection of this inhibitor alone (7-NI+VEH), in the absence of PRE, did not affect spinal Nox2 activation in comparison to the VEH+VEH group.
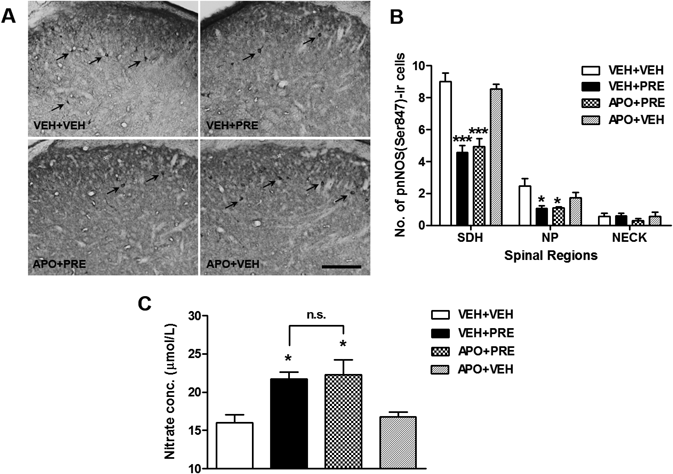
Sig-1R-Induced Nox Activation Has No Effect on the Number of pnNOS-ir Cells and NO Concentration in the Spinal Cord Dorsal HornSince phosphorylation of nNOS (pnNOS) at serine 847 (Ser847) inhibits nNOS activity,5,6) a decrease in the pnNOS expression is considered to reflect an increase in the active forms of nNOS. The changes in the expression of pnNOS in the spinal cord dorsal horn were measured by using an immmunohistochemistry. Intrathecal injection of PRE084 (3 nmol, VEH+PRE) significantly decreased the phosphorylation of nNOS (as evidenced by a decrease in pnNOS-ir cells) in the SDH (laminae I–II) and NP regions (laminae III–IV) of the lumbar spinal cord dorsal horn (Figs. 4A, B; * p<0.05, *** p<0.001 vs. VEH+VEH). This decrease was not blocked by i.t. pretreatment with the Nox inhibitor, apocynin (100 nmol, APO+PRE) (* p<0.05, *** p<0.001 vs. VEH+VEH). The i.t. injection of this inhibitor alone (APO+VEH), in the absence of PRE, did not affect the number of pnNOS-ir cells in comparison to the VEH+VEH group. We further confirmed changes in spinal NO production by examining changes in nitrate concentration. PRE084 (3 nmol, VEH+PRE) significantly increased total NO levels as compared with that of the vehicle-treated group, while this increase was not suppressed by pretreatment with APO (Fig. 4C; * p<0.05 vs. VEH+VEH, non-significance vs. VEH+PRE).
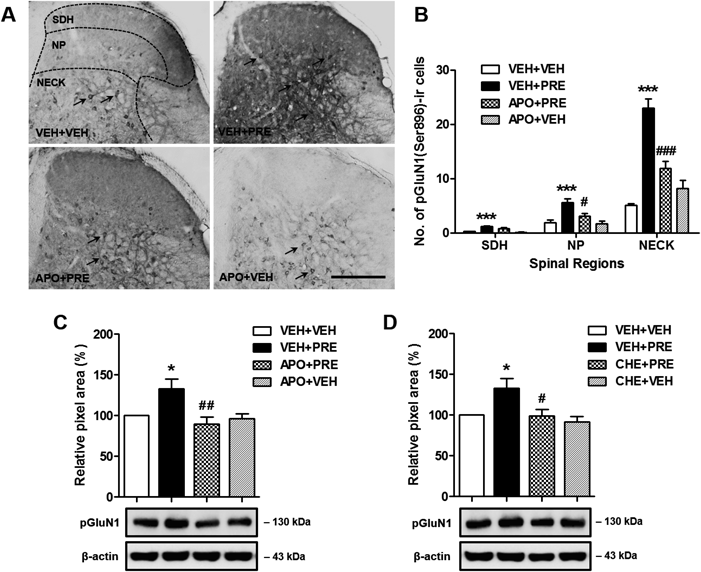
Sig-1R-Induced Nox Activation Increases pGluN1 Expression in the Spinal Cord Dorsal HornIntrathecal administration of PRE084 (3 nmol, VEH+PRE) significantly increased the number of cells in the SDH (laminae I–II), NP (laminae III–IV) and NECK regions (laminae V–VI) of the lumbar spinal cord dorsal horn that were immunoreactive for PKC-dependent phosphorylation (at the Ser896 site) of the NMDA receptor GluN1 subunit (pGluN1) (Figs. 5A, B; *** p<0.001 vs. VEH+VEH). Intrathecal pretreatment with the Nox inhibitor, apocynin (100 nmol, APO+PRE) prior to PRE084 administration, dramatically suppressed this PRE084-induced increase in the number of PKC-dependent pGluN1-ir cells in the spinal cord dorsal horn, particularly in the NP and NECK regions (Figs. 5A, B; # p<0.05, ### p<0.001 vs. VEH+PRE). The i.t. injection of this inhibitor alone (APO+VEH), in the absence of PRE, did not affect spinal pGluN1 expression in comparison to the VEH+VEH group. In addition, the effect of APO and the PKC inhibitor, chelerythrine (CHE) on PRE084-induced change in the pGluN1 expression was confirmed by Western blot analysis. As illustrated in Figs. 5C and D, APO (100 nmol, APO+PRE) or CHE (1 nmol, CHE+PRE) pretreatment significantly reduced PRE084-induced increase in PKC-dependent pGluN1 expression (Figs. 5C, D; * p<0.05 vs. VEH+VEH, # p<0.05, ## p<0.01 vs. VEH+PRE).
DISCUSSION
It is well recognized that Sig-1Rs are predominantly localized to the endoplasmic reticulum in cells of the nervous system and play an important role in a variety of cellular signaling processes via modulation of intracellular Ca2+ levels.22,23) Previous studies from our laboratory have shown that activation of spinal Sig-1Rs is critical for the inflammatory and neuropathic pain induced by formalin injection and chronic sciatic nerve injury, respectively.24,25) These nociceptive effects of Sig-1Rs are associated with activation of Ca2+ dependent second messenger cascades via a phospholipase C-inositol triphosphate (PLC-IP3)-PKC signaling pathway leading to the potentiation of NMDA receptor activity via increases in PKC-dependent phosphorylation of the NMDA receptor GluN1 subunit (pGluN1).14,26) In the present study, i.t. administration of the Sig-1R agonist, PRE084 was shown to induce both mechanical and thermal hypersensitivity in mice. I.t. pretreatment with either the nNOS inhibitor, 7-NI or the Nox inhibitor, apocynin attenuated this pain hypersensitivity. Since NO and superoxide (O2−) can combine to form the powerful and toxic oxidant, peroxynitrite anion (ONOO−),27) we examined whether peroxynitrite mediates the nociceptive effects induced by PRE084 administration. However, we found that i.t. administration of the peroxynitrite scavenger, FeTPPS had no effect on Sig-1R-induced pain hypersensitivity. These results indicate that spinal Sig-1R activation induces pain hypersensitivity via modulation of nNOS and Nox activation, while the convergence of NO and superoxide to produce peroxynitrite does not affect Sig-1R-induced nociception.
In the present study, Sig-1R activation was shown to significantly increase NO production in the lumbar spinal cord dorsal horn. It is well known that NO is synthesized in biological systems by three NOS enzymes (nNOS, iNOS, or eNOS).1) Importantly, we demonstrate that only nNOS inhibition caused a significant reduction in the Sig-1R agonist-induced increase in spinal NO production, whereas iNOS and eNOS inhibition had no effect on this PRE084-induced increase in NO production. Furthermore, we recently demonstrated that activation of spinal nNOS is mechanistically associated with the PRE084-induced increase in paw pinch-evoked spinal cord Fos expression as well as in PKC-dependent pGluN1 expression.6) Thus, we conclude that the Sig-1R-induced increase in NO production is most closely related to nNOS activation in the spinal cord. Since Sig-1Rs are abundant in astrocytes within the superficial dorsal horn of the spinal cord,25) it is likely that activation of Sig-1R may indirectly increase nNOS activity. In this regard, we have recently demonstrated that Sig-1R activation modulates the production of D-serine in spinal astrocytes, which contributes to the development of mechanical allodynia in a mouse model of neuropathic pain.28) Since D-serine can modulate NMDA receptor activity as an endogenous ligand for the glycine site of this receptor,29) it is possible that D-serine released from astrocytes can regulate nNOS activity in nearby neurons. However, this indirect effect of Sig-1R activation on nNOS activity needs to be confirmed by further investigation.
Several reports have suggested that nNOS-derived NO stimulates Nox2 activation, which then links NMDA receptor activation with Nox2-dependent ROS production.9,30) These findings raise the possibility of a relationship between nNOS and Nox2 activation that is induced by stimulation of spinal Sig-1Rs. Accordingly we investigated whether activation of spinal Sig-1Rs and consequent nNOS-derived NO is associated with Nox2 activation in the spinal cord leading to an increase in the production of ROS. An increase in the membrane fraction of the Nox2 subunit p47phox has been considered to represent an increase in the active form of the Nox2 enzyme.10) Based on this assumption, we assessed the expression of p47phox in the membrane fraction as a specific indicator of Nox2 activation. In addition, it has been shown that phosphorylation of nNOS (pnNOS) at Ser847 reduces nNOS activity by inhibiting Ca2+/CaM binding, which is a critical step in nNOS activation.5) In this regard, an observed decrease in pnNOS possibly represents an increase in nNOS activity. In the present study, we found that pretreatment with the nNOS inhibitor, 7-NI reduces the PRE084-induced increase in Nox2 activity and ROS production in the superficial dorsal horn (SDH, laminae I–II) of lumbar spinal cord. However, administration of the Nox inhibitor, apocynin did not modify the PRE084-induced change in pnNOS expression. Since nociceptive primary afferent neurons synapse with second order neurons mainly in lamina I and II of spinal dorsal horn,31) these results suggest the possibility that Sig-1R-induced Nox2 activation in the SDH region plays an important role in modulating spinal nociceptive transmission from the periphery to the CNS as a downstream signaling component of the nNOS-NO signaling cascade.
A number of recent reports suggest several possible mechanisms by which an increase in NO induces Nox2 activation. Based on in vitro results using cortical neuronal cultures, Girouard et al. suggested that the effect of NO on ROS production induced by NMDA receptor activation requires soluble guanylyl cyclase (sGC), cGMP and protein kinase G (PKG) activation.9) PKG can activate the small GTPase Rac1 supporting the induction of functional active Nox enzyme assembly.32,33) These findings suggest that NO-sGC-PKG signaling cascades may have pro-oxidant effects in the CNS via modulation of Nox activity and concomitant ROS production, which could serve to modulate persistent pain.7) On the other hand, there are several reports suggesting that NO signaling can produce analgesia.34) Activation of the NO-cGMP pathway in the periphery causes antinociception via opening ATP-sensitive K+ channels with a consequent increase in the K+ current, resulting in nociceptor desensitization.35) In addition, i.t. administration of a cGMP analog has been shown to either inhibit or facilitate synaptic transmission of nociceptive stimuli in the spinal cord depending on the dose.36) This dual effect of NO may result from distinct intracellular signaling pathways, which are dependent on the experimental models used for pain evaluation, on the type and phase of the nociceptive process, and on the site of activation.1,36) These differences require further investigation to clarify the role of NO in the underlying pathophysiological mechanisms of pain.
There is now considerable evidence that spinal NMDA receptors play a critical role in the process of ‘central sensitization,’ an essential phenomenon in the development of chronic pain.37,38) Phosphorylation of the NMDA receptor GluN1 subunit at serine residues increases the excitability and synaptic efficacy of neurons in spinal pain pathways by reducing the Mg2+ block and by increasing the probability of channel openings.39) In the present study, i.t. administration of the Sig-1R agonist, PRE084 results in a significant increase in the number of PKC-dependent pGluN1 (at Ser896 site)-ir cells in the lumbar spinal cord dorsal horn. This increase is significantly reduced by the pretreatment with the Nox inhibitor, apocynin. These results suggest that Sig-1R-induced Nox2 activation and the accompanying ROS production can modulate PKC activity, resulting in potentiation of NMDA receptor function via an increase in the phosphorylation of GluN1 (at Ser896 site). ROS are known to modify diverse intracellular signaling pathways including protein phosphatases, protein kinases, and transcription factors.40) In particular, superoxide stimulates autonomous PKC activity via thiol oxidation in hippocampal neurons.41,42) In addition, hydrogen peroxide can enhance the activity of PKC including its α and ε isoforms, which is reported to associate with pGluN1 at the Ser896 site,26) by increasing tyrosine phosphorylation of PKC and inhibiting protein tyrosine phosphatase activity.43–45) These studies suggest the possibility that ROS derived from Nox2 activation can increase phosphorylation of the NMDA receptor GluN1 subunit via both direct and indirect modulation of PKC activity.
In the present study, the differential distribution of pnNOS-ir cells, DHE staining (which represents ROS production), and pGluN1-ir cells in the spinal cord dorsal horn raises an important mechanistic question to consider. Since NO is a diffusible gas and readily permeates cell membranes, it can diffuse from its site of production to act on other adjacent cells leading to stimulation of ROS production in the spinal cord.1) While ROS production was increased in the SDH region (laminae I–II) of the spinal cord, most of the PKC-dependent pGluN1-ir cells modulated by ROS were detected in the deeper dorsal horn (laminae III–IV and V–VI). One explanation for this discrepancy might be suggested from the findings of Simone et al. showing that capsaicin-induced C-fiber stimulation increases the responses not only from SDH neurons, but also from many deep dorsal horn neurons.7,46) It has been also demonstrated that many deep dorsal horn neurons express neurokinin-1 receptors and have dorsal dendrites that project to the superficial dorsal horn.47) Moreover, inhibitory interneurons in the dorsal horn can reduce the excitability of spinal projection neurons in the deeper layers and subsequently attenuate the inputs from nociceptive afferent fibers.48) This anatomical framework is further supported by studies suggesting that ROS can reduce spinal γ-aminobutyric acid (GABA) release and enhance responses of the spinal wide dynamic range neurons.49,50) Thus, even though ROS is primarily produced in the SDH by Sig-1R-induced nNOS activation, it may affect NMDA receptors located on deeper dorsal horn cells that convey nociceptive information to higher levels of the central nervous system.
It is well known that the responses of primary sensory neurons to mechanical and thermal stimulation are different. There are several studies suggesting that depletion of capsaicin sensitive primary afferents using an analog of capsaicin, resiniferatoxin (RTX), suppressed thermal hyperalgesia, but not mechanical allodynia, in several neuropathic pain models.51,52) In addition, Roh et al. demonstrated that peripheral nerve injury increases the number of pGluN1-ir neurons in the spinal cord dorsal horn, and this increase is significantly attenuated in the SDH (laminae I–II) and NECK (laminae V–VI), but not the NP (laminae III–IV), regions of the dorsal horn in RTX-pretreated neuropathic rats.53) These studies suggest that the development of thermal hyperalgesia is related to increase in pGluN1 expression in the SDH and NECK regions of the dorsal horn. In contrast, low-threshold mechanoreceptive and wide dynamic range neurons are mainly distributed in the NP and NECK regions, and these neurons are involved in the signal transmission of tactile information in normal animals.53) In this regard, it may be possible that PRE084-induced mechanical hypersensitivity is related to an increase in pGluN1 expression in the NP and NECK regions of the dorsal horn, and PRE084-induced thermal hypersensitivity is related to an increase in pGluN1 expression in the SDH and NECK regions of the dorsal horn. However, further investigations considering the pathophysiological and anatomical evidence are needed to clarify the mechanisms of mechanical and thermal hypersensitivity associated with a variety of acute and chronic pain conditions.
In conclusion, the present study demonstrates that nNOS-derived NO signaling modulated by spinal Sig-1R activation increases Nox2 activity and concomitant ROS production, which leads to a ROS-induced increase in PKC-dependent pGluN1 expression in the spinal cord dorsal horn and the development of pain hypersensitivity. These findings suggest that the downstream NO signaling cascade contributes to the increase in ROS production, which plays an important role in the development of spinal Sig-1R-mediated pain hypersensitivity. Clearly increased understanding of this interaction is critical to expanding our knowledge of the role played by NO in Sig-1R-dependent neuropathy.
Acknowledgment
This research was supported by a Grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant No.: HI15C2589010015).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Cury Y, Picolo G, Gutierrez VP, Ferreira SH. Pain and analgesia: The dual effect of nitric oxide in the nociceptive system. Nitric Oxide, 25, 243–254 (2011).
- 2) Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem., 274, 27467–27473 (1999).
- 3) Ferreira J, Santos AR, Calixto JB. The role of systemic, spinal and supraspinal L-arginine–nitric oxide–cGMP pathway in thermal hyperalgesia caused by intrathecal injection of glutamate in mice. Neuropharmacology, 38, 835–842 (1999).
- 4) Kawamata T, Omote K. Activation of spinal N-methyl-D-aspartate receptors stimulates a nitric oxide/cyclic guanosine 3,5-monophosphate/glutamate release cascade in nociceptive signaling. Anesthesiology, 91, 1415–1424 (1999).
- 5) Rameau GA, Chiu LY, Ziff EB. Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-D-aspartate receptor. J. Biol. Chem., 279, 14307–14314 (2004).
- 6) Roh DH, Choi SR, Yoon SY, Kang SY, Moon JY, Kwon SG, Han HJ, Beitz AJ, Lee JH. Spinal neuronal NOS activation mediates sigma-1 receptor-induced mechanical and thermal hypersensitivity in mice: involvement of PKC-dependent GluN1 phosphorylation. Br. J. Pharmacol., 163, 1707–1720 (2011).
- 7) Gao X, Kim HK, Chung JM, Chung K. Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain, 131, 262–271 (2007).
- 8) Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev., 87, 245–313 (2007).
- 9) Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci., 29, 2545–2552 (2009).
- 10) Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci., 12, 857–863 (2009).
- 11) Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol., 4, 181–189 (2004).
- 12) DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J. Leukoc. Biol., 60, 677–691 (1996).
- 13) Choi SR, Roh DH, Yoon SY, Kang SY, Moon JY, Kwon SG, Choi HS, Han HJ, Beitz AJ, Oh SB, Lee JH. Spinal sigma-1 receptors activate NADPH oxidase 2 leading to the induction of pain hypersensitivity in mice and mechanical allodynia in neuropathic rats. Pharmacol. Res., 74, 56–67 (2013).
- 14) Roh DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Kim KW, Han HJ, Beitz AJ, Lee JH. Intrathecal administration of sigma-1 receptor agonists facilitates nociception: involvement of a protein kinase C-dependent pathway. J. Neurosci. Res., 86, 3644–3654 (2008).
- 15) National institute of Health, Guide for the Care and Use of Laboratory Animals. DHEW Publication (NIH). 2nd revised ed. Bethesda, MD, Office of Science and Health Reports, DRR/NIH (1985).
- 16) Yeo JF, Ling SF, Tang N, Ong WY. Antinociceptive effect of CNS peroxynitrite scavenger in a mouse model of orofacial pain. Exp. Brain Res., 184, 435–438 (2008).
- 17) Yoon SY, Roh DH, Seo HS, Kang SY, Moon JY, Song S, Beitz AJ, Lee JH. An increase in spinal dehydroepiandrosterone sulfate (DHEAS) enhances NMDA-induced pain via phosphorylation of the NR1 subunit in mice: involvement of the sigma-1 receptor. Neuropharmacology, 59, 460–467 (2010).
- 18) Lee JS, Zhang Y, Ro JY. Involvement of neuronal, inducible and endothelial nitric oxide synthases in capsaicin-induced muscle hypersensitivity. Eur. J. Pain, 13, 924–928 (2009).
- 19) Doyle T, Bryant L, Muscoli C, Cuzzocrea S, Esposito E, Chen Z, Salvemini D. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci. Lett., 483, 85–89 (2010).
- 20) Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur. J. Pharmacol., 67, 313–316 (1980).
- 21) Abbadie C, Besson JM. Chronic treatments with aspirin or acetaminophen reduce both the development of polyarthritis and Fos-like immunoreactivity in rat lumbar spinal cord. Pain, 57, 45–54 (1994).
- 22) Monnet FP. Sigma-1 receptor as regulator of neuronal intracellular Ca2+: clinical and therapeutic relevance. Biol. Cell, 97, 873–883 (2005).
- 23) Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci., 31, 557–566 (2010).
- 24) Roh DH, Yoon SY. Sigma-1 receptor antagonist, BD1047 reduces nociceptive responses and phosphorylation of p38 MAPK in mice orofacial formalin model. Biol. Pharm. Bull., 37, 145–151 (2014).
- 25) Moon JY, Roh DH, Yoon SY, Choi SR, Kwon SG, Choi HS, Kang SY, Han HJ, Beitz AJ, Oh SB, Lee JH. σ1 receptors activate astrocytes via p38 MAPK phosphorylation leading to the development of mechanical allodynia in a mouse model of neuropathic pain. Br. J. Pharmacol., 171, 5881–5897 (2014).
- 26) Roh DH, Yoon SY, Seo HS, Kang SY, Moon JY, Song S, Beitz AJ, Lee JH. Sigma-1 receptor-induced increase in murine spinal NR1 phosphorylation is mediated by the PKCalpha and epsilon, but not the PKCzeta, isoforms. Neurosci. Lett., 477, 95–99 (2010).
- 27) Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol., 271, C1424–C1437 (1996).
- 28) Moon JY, Choi SR, Roh DH, Yoon SY, Kwon SG, Choi HS, Kang SY, Han HJ, Kim HW, Beitz AJ, Oh SB, Lee JH. Spinal sigma-1 receptor activation increases the production of D-serine in astrocytes which contributes to the development of mechanical allodynia in a mouse model of neuropathic pain. Pharmacol. Res., 100, 353–364 (2015).
- 29) Mothet JP, Parent AT, Wolosker H, Brady RO Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A., 97, 4926–4931 (2000).
- 30) Little JW, Doyle T, Salvemini D. Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain. Amino Acids, 42, 75–94 (2012).
- 31) Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci., 11, 823–836 (2010).
- 32) Hou Y, Ye RD, Browning DD. Activation of the small GTPase Rac1 by cGMP-dependent protein kinase. Cell. Signal., 16, 1061–1069 (2004).
- 33) Gorzalczany Y, Sigal N, Itan M, Lotan O, Pick E. Targeting of Rac1 to the phagocyte membrane is sufficient for the induction of NADPH oxidase assembly. J. Biol. Chem., 275, 40073–40081 (2000).
- 34) Durate ID, Lorenzetti BB, Ferreira SH. Peripheral analgesia and activation of the nitric oxide-cyclic GMP pathway. Eur. J. Pharmacol., 186, 289–293 (1990).
- 35) Sachs D, Cunha FQ, Ferreira SH. Peripheral analgesic blockade of hypernociception: activation of arginine/NO/cGMP/protein kinase G/ATP-sensitive K+ channel pathway. Proc. Natl. Acad. Sci. U.S.A., 101, 3680–3685 (2004).
- 36) Tegeder I, Schmidtko A, Niederberger E, Ruth P, Geisslinger G. Dual effects of spinally delivered 8-bromo-cyclic guanosine mono-phosphate (8-bromo-cGMP) in formalin-induced nociception in rats. Neurosci. Lett., 332, 146–150 (2002).
- 37) Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet, 353, 1959–1964 (1999).
- 38) Ultenius C, Linderoth B, Meyerson BA, Wallin J. Spinal NMDA receptor phosphorylation correlates with the presence of neuropathic signs following peripheral nerve injury in the rat. Neurosci. Lett., 399, 85–90 (2006).
- 39) Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature, 356, 521–523 (1992).
- 40) Maher P, Schubert D. Signaling by reactive oxygen species in the nervous system. Cell. Mol. Life Sci., 57, 1287–1305 (2000).
- 41) Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J. Biol. Chem., 273, 4516–4522 (1998).
- 42) Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J. Biol. Chem., 275, 24136–24145 (2000).
- 43) Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl. Acad. Sci. U.S.A., 94, 11233–11237 (1997).
- 44) Sullivan SG, Chiu DTY, Errasfa M, Wang JM, Qi JS, Stern A. Effects of H2O2 on protein tyrosine phosphatase activity in her14 cells. Free Radic. Biol. Med., 16, 399–403 (1994).
- 45) Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry, 37, 5633–5642 (1998).
- 46) Simone DA, Sorkin LS, Oh U, Chung JM, Owens C, LaMotte RH, Willis WD. Neurogenic hyperalgesia: central neural correlates in responses of spinothalamic tract neurons. J. Neurophysiol., 66, 228–246 (1991).
- 47) Naim M, Spike RC, Watt C, Shehab SA, Todd AJ. Cells in laminae III and IV of the rat spinal cord that possess the neurokinin-1 receptor and have dorsally directed dendrites receive a major synaptic input from tachykinin-containing primary afferents. J. Neurosci., 17, 5536–5548 (1997).
- 48) Prager JP. What does the mechanism of spinal cord stimulation tell us about complex regional pain syndrome? Pain Med., 11, 1278–1283 (2010).
- 49) Yowtak J, Lee KY, Kim HY, Wang J, Kim HK, Chung K, Chung JM. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain, 152, 844–852 (2011).
- 50) Lee I, Kim HK, Kim JH, Chung K, Chung JM. The role of reactive oxygen species in capsaicin-induced mechanical hyperalgesia and in the activities of dorsal horn neurons. Pain, 133, 9–17 (2007).
- 51) Ossipov MH, Bian D, Malan TP Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain, 79, 127–133 (1999).
- 52) Kingery WS, Agashe GS, Guo TZ, Davies MF, Clark JD, Maze M. Capsaicin sensitive afferents mediate the development of heat hyperalgesia and hindpaw edema after sciatic section in rats. Neurosci. Lett., 318, 39–43 (2002).
- 53) Roh DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Han HJ, Beitz AJ, Lee JH. Depletion of capsaicin-sensitive afferents prevents lamina-dependent increases in spinal N-methyl-D-aspartate receptor subunit 1 expression and phosphorylation associated with thermal hyperalgesia in neuropathic rats. Eur. J. Pain, 12, 552–563 (2008).