RESULTS AND DISCUSSION
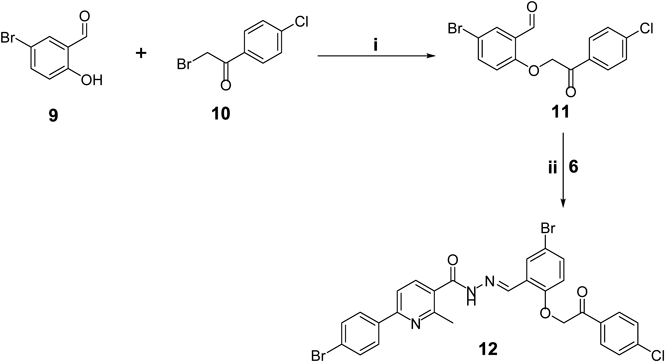
Discussion of ChemistryThe synthetic strategy for preparing the target nicotinic acid hydrazides 8a–g, 12 and 16a, b was illustrated in Charts 1–3. In Chart 1, synthesis of target hydrazides required three successive reactions. The enaminone 3 reacted with ethyl acetoacetate 4 and ammonium acetate in hot glacial acetic acid, to furnish the ethyl 6-(4-bromophenyl)-2-methylnicotinate 5 in a three-component heterocyclocondensation process. This was followed by hydrazinolysis of the later ester 5 to yield the nicotinic acid hydrazide 6, in 90% yield. Condensation of the hydrazide 6 with different aldehydes 7a–g, in ethyl alcohol in the presence of a catalytic amount of acetic acid furnished the target compounds 8a–g (Chart 1).

IR spectra of compounds 8a–g revealed the appearance of bands around 1660 and 3170 cm−1 assigned to the carbonyl and NH groups, respectively. Also, 1H-NMR spectra of 8a–g displayed a singlet D2O-exchangeable signal attributable to NH proton of hydrazine function (=N–NH–) around δ 12 ppm, while the methyl (–CH3) and methine (–CH=N–) protons appeared around δ 2.60 and 8.40 ppm, respectively, as singlet signals. Also, additional singlet signals corresponding to methoxy protons resonating around δ 3.70 ppm in case of 8c and d were observed. Furthermore, 13C-NMR showed the characteristic signals for the carbons of carbonyl groups around δ 170.30 ppm, whereas the carbons for the methyl groups appeared around δ 23.40 ppm.
IR spectrum of 12 confirmed the presence of two peaks at 1701 and 3155 cm−1 corresponding to the (C=O) and NH groups, respectively. The 1H-NMR spectrum of 12 showed two singlet signals of the aliphatic (–OCH2–) and methine (–CH=N–) protons at δ 5.72 and 8.00 ppm, respectively, also, 1H-NMR spectrum revealed the presence of D2O exchangable NH proton at a δ 12.14 ppm.
Finally, p-flourobenzaldehyde 13 was reacted with an appropriate alicyclic secondary amine 14a, b in dry dimethyl sulfoxide (DMSO) in presence of potassium carbonate to furnish aldehydes 15a, b (yield, 65 and 70%, respectively), which condensed with hydrazide 6 in absolute ethyl alcohol and catalytic amount of glacial acetic acid to afford the respective target derivative 16a, b in good yield (Chart 3).
1H-NMR spectra for the latter products showed singlet D2O-exchangeable signals attributable to NH proton of the hydrazine function (=N–NH–) around δ 11.70 ppm, while the methyl (–CH3) protons appeared at δ 2.65 ppm as singlet signals. Also, their 1H-NMR revealed the protons of morpholine and piperidine moiety in the aliphatic regions; (δ 1.53–1.59 and 3.19 ppm for piperidine moiety) and (δ 3.11 and 3.367 ppm for morpholine moiety moiety).
The prepared hydrazones in this study appeared in the 1H-NMR spectra (in the solution state, DMSO-d6) as mixed E and Z geometrical isomers due to the imine C=N double bond. While, the X-ray single crystal analysis for compound 8a illustrates the interesting property of such hydrazone in the crystalline state as an E geometrical isomer around (C=N) (Supplementary Materials).33–35)
Discussion of Biological EvaluationAnti-microbial ActivityAntimycobacterial, antifungal and antibacterial activities for the target compounds 8a–g, 12, 16a, b and reference drugs were in vitro evaluated at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Cairo, Egypt. The results are represented in Tables 1–3.
Table 1. Structures, Antitubercular Activities, log
P Measurements and Drug-Likeness Model Score of the Synthesized Derivatives
a: Calculated by [www.molinspiration.com]; b: Calculated by ref. 48; NA: No activity (>150 µg/mL).
Table 2. Antifungal Activity of the Tested Standards and Synthesized Compounds; Expressed as Inhibition Diameter Zones (I.Z.) in mm, and Minimum Inhibitory Concentrations (MIC) in µg/mL
 |
|---|
| Compound | R/Ar/X | I.Z. (mm) | MIC (µg/mL) |
|---|
| Af | Ca | Af | Ca |
|---|
| 8a | 2,4-(Cl2)-C6H3- | 18.2±0.38 | 17.3±0.58 | 7.81 | 15.63 |
| 8b | 2,6-(Cl2)-C6H3- | 23.4±0.25 | 24.1±1.5 | 0.98 | 0.98 |
| 8c | 3,4-(OCH3)2-C6H3- | 21.2±0.58 | 18.3±0.58 | 1.95 | 7.81 |
| 8d | 2,5-(OCH3)2-C6H3- | 24.7±0.38 | 20.8±1.2 | 0.49 | 1.95 |
| 8e | 5-Br-2-OH-C6H3- | 20.1±0.63 | 18.3±0.58 | 62.5 | 125 |
| 8f | Thiophen-2-yl | 18.2±0.58 | 15.4±0.34 | 7.81 | 31.25 |
| 8g | Pyridin-3-yl | 22.9±0.25 | 19.3±1.53 | 0.98 | 3.9 |
| 12 | — | 13.6±0.37 | 11.4±1.20 | 3.9 | 7.81 |
| 16a | O | 14.2±0.25 | 13.6±1.25 | 62.5 | 62.5 |
| 16b | CH2 | 16.9±0.58 | 16.4±1.33 | 15.63 | 31.25 |
| AB | | 23.1±0.90 | 21.3±0.72 | 0.98 | 1.95 |
The screening organisms, Aspergillus fumigatus (RCMB 02568, Af), Candida albicans (RCMB 05036, Ca), AB: Amphotericin B.
Table 3. Antibacterial Activity of the Tested Standards and Synthesized Compounds; Expressed as Inhibition Diameter Zones (I.Z.) in mm, and Minimum Inhibitory Concentrations (MIC) in µg/mL
| Comp. | Gram-positive bacteria | Gram-negative bacteria |
|---|
| I.Z. (mm) | MIC (µg/mL) | I.Z. (mm) | MIC (µg/mL) |
|---|
| Sa | Sp | Bs | Sa | Sp | Bs | Pa | St | Kp | Ec | Pa | St | Kp | Ec |
|---|
| 8a | 22.4 | 21.3 | 20.6 | 0.98 | 1.95 | 1.95 | 18.3 | 21.3 | 22.4 | 22.6 | 7.81 | 1.95 | 0.98 | 0.98 |
| 8b | 25.2 | 24.1 | 26.4 | 0.49 | 0.49 | 0.24 | 21.3 | 22.7 | 25.4 | 26.3 | 1.95 | 0.98 | 0.49 | 0.49 |
| 8c | 21.3 | 22.1 | 20.3 | 1.95 | 0.98 | 3.9 | NA | 18.4 | 20.3 | 21.2 | NA | 7.81 | 3.9 | 1.95 |
| 8d | 20.4 | 22.6 | 19.8 | 3.9 | 0.98 | 3.9 | 21.3 | 23.4 | 24.9 | 26.4 | 1.95 | 0.98 | 0.49 | 0.49 |
| 8e | 18.7 | 21.3 | 20.2 | 7.81 | 7.81 | 31.25 | NA | 18.3 | 21.2 | 21.9 | NA | 31.25 | 15.63 | 7.81 |
| 8f | 13.6 | 16.4 | 12.4 | 62.5 | 31.25 | 125 | NA | 13.2 | 15.7 | 17.3 | NA | 62.5 | 31.25 | 15.63 |
| 8g | 24.2 | 23.6 | 21.3 | 0.49 | 0.49 | 1.95 | 19.6 | 22.4 | 24.6 | 25.1 | 3.9 | 0.98 | 0.49 | 0.49 |
| 12 | 18.2 | 17.6 | 16.1 | 3.9 | 1.95 | 3.9 | NA | 15.6 | 17.1 | 17.9 | NA | 7.81 | 1.95 | 62.5 |
| 16a | 17.3 | 17.9 | 13.6 | 15.63 | 7.81 | 62.5 | NA | 11.2 | 13.4 | 16.2 | NA | 125 | 62.5 | 31.25 |
| 16b | 21.9 | 21.2 | 20.1 | 0.98 | 1.95 | 3.9 | 16.3 | 20.4 | 21.6 | 22.2 | 31.25 | 3.9 | 0.98 | 0.98 |
| Ap | 26.4 | 25.3 | 28.9 | 0.98 | 0.49 | 0.49 | | | | | | | | |
| GM | | | | | | | 26.3 | 23.2 | 27.5 | 27.3 | 0.98 | 0.98 | 0.49 | 0.49 |
| CF | 22.2 | 23.3 | 25.4 | 1.95 | 1.95 | 1.95 | 23.1 | 21.8 | 24.7 | 24.3 | 3.90 | 3.90 | 3.90 | 1.95 |
NA: No Activity. The screening organisms, Gram-positive bacteria: Staphylococcus aureus (RCMB 010028, Sa), Streptococus pneumoniae (RCMB 010010, Sp), and Bacillus subtilis (RCMB 010069, Bs). Gram-negative bacteria: Pseudomonas aeruginosa (RCMB 010043, Pa), Salmonella typhimurium (RCMB 010315, St), Klebsiella penumoniae (RCMB 0010093, Kp) and Escherichia coli (RCMB 010052, Ec), AP: Ampicillin, GM: Gentamicin, CF: Ciprofloxacin.
Target compounds 8a–g, 12, 16a, b were in vitro evaluated for their antitubercular activity against M. tuberculosis (RCMB 010126) using the microplate Alamar blue assay (MABA).36) Isoniazide and pyrazinamide were used as reference drugs. The results of the in vitro antimycobacterial screening, as percent inhibition and minimum inhibitory concentration (MIC), are summarized in Table 1. Among the tested hydrazones, compound 8b emerged as the most potent member with significant antimycobacterial activity with a MIC value of 3.9 µg/mL. Compound 8g also exhibited moderate activity (MIC=15.63 µg/mL). Otherwise, compounds 8c, 8d and 12 possessed modest anti-tubercular activity with MIC values range of: 62.5 to 125 µg/mL.
Upon examining the MIC results in Table 1, worthy information about the structure activity relationships, of compounds 8a–g, 12, 16a, b with respect to their antitubercular activity, could be concluded. Firstly, we examined the effect of grafting different two substituents on the pendent phenyl ring on the prepared hydrazones activity. Incorporation of two lipophilic groups as chloro or methoxy groups led to compounds 8b–d with potent to modest activity (MIC: 3.90–100 µg/mL). Conversely, introduction of hydrophilic group (–OH) and lipophilic one (–Br), as in compound 8e, abolished the activity. Also, appending alicyclic secondary amines; morpholine or piperidine as in compounds 16a and b, diminished the activity, suggesting that substitution of the pendant phenyl group with two lipophilic groups is crucial for the antimycobacterial activity.
On the other hand, the positional isomerism; moving the two chlorine atoms from 2,4 positions in compound 8a to 2,6 positions in 8b, resulted in about 25 fold increase in the antimycobacterial activity, hinting that the positions of such two lipophilic groups greatly affect the activity. The order of activity of the disubstituted hydrazones 8a–d was 2,6-dichloro 8b>3,4-dimethoxy 8c>2,4-dichloro 8a and 2,5-dimethoxy 8d (MIC=3.90, 62.5, 100 and 100 µg/mL, respectively). Moreover, extending compound 8e (MIC=>150 µg/mL) with the large lipophilic 4-chlorophenyl moiety through ketomethylene (–CH2–CO–) linker, led to a comparatively active counterpart 12 (MIC=125 µg/mL), confirming that presence of hydrophilic group is not favorable for activity.
Finally, we examined the effect of bioisosteric replacement of the pendant substituted phenyl moiety in compounds 8a–d. Interestingly, attaching 3-pyridyl group, compound 8g, as bioisostere to the phenyl one maintained the activity (MIC=62.5 µg/mL), while 2-thienyl group, 8f, abolished the activity.
Anti-fungal ActivityTarget compounds 8a–g, 12, 16a, b and amphotericin, a membrane-active polyene macrolide antibiotic and an antifungal reference drug, were in vitro evaluated for their antifungal activity, by inhibition zone (I.Z.) technique and MIC,32) towards Aspergillus fumigatus and Candida albicans, Table 2.
Aspergillus species, globally ubiquitous saprophytes, are found in a variety of ecological niches. More than 200 species of aspergilli have been identified, less than 20 of which are recognized to cause human disease. Among them, A. fumigatus was found to be the most prevalent and is predominantly responsible for the increased incidence of invasive aspergillosis in the immunocompromised patient.
As for the antifungal activity, data in Table 2 showed that the 2,5-dimethoxybenzylidene derivative 8d demonstrated the best activity amongst the tested compounds against A. fumigatus (MIC=0.49 µg/mL), which was 2-fold more potent than the reference dug, amphotericin (MIC=0.98 µg/mL). While, compounds 8b and g were equipotent to amphotericin against A. fumigatus. Furthermore, derivatives 8a, c, f, 12 and 16b showed moderate activity against A. fumigatus (MIC=7.81, 1.95, 7.81, 3.9 and 15.63 µg/mL, respectively)
Regarding the activity against Candida albicans, the 2,6-dichlorobenzylidene counterpart 8b was the most active one with MIC=0.98 µg/mL. Also, compound 8d displayed good activity and was equipotent to amphotericin against Candida albicans, (MIC=1.95 µg/mL). Moreover, compounds 8a, c, f, g, 12 and 16b were moderately active, against Candida albicans, with MIC values ranging from 3.9 to 31.25 µg/mL.
Antibacterial ActivityThe results in Table 3 showed that the compound 8b demonstrated broad and excellent activity among all the tested compounds, it was twice the activity of ampicillin against Staphylococcus aureus and Bacillus subtilis (0.49, 0.24 µg/mL, ampicillin, 0.98, 0.49 µg/mL), it was also equipotent to Gentamicin against Salmonella typhimurium, Klebsiella penumoniae and Escherichia coli (0.98, 0.49, 0.49 µg/mL). The (2,4-dichlorobenzylidene)-2-methylnicotinohydrazide 8a was equipotent to ampicillin against S. aureus, (0.98 µg/mL), it also possessed the same activity against Streptococus pneumoniae and B. subtilis as ciprofloxacin (1.95 µg/mL). As for the Gram-negative (g−ve) activity, it exhibited 4-foldsthe activity of ciprofloxacin against K. penumoniae and possessed twice its activity against S. typhimurium and E. coli. On the other hand, compound 8g demonstrated twice the activity of ampicillin against S. aureus, the same potency as ampicillin against (S. pneumoniae) as well as (the effect of gentamicin on) the g−ve organisms, S. typhimurium, K. penumoniae and E. coli. Furthermore, the benzylidene derivative 8c showed equipotent activity to ciprofloxacin against S. aureus, K. penumoniae, E. coli and was almost 2-folds its activity against S. pneumoniae. Regarding the activity of piperidinylbenzylidene counterpart 16b, it exhibited g−ve and Gram-positive (g+ve) activity, it was found to demonstrate the same activity as ampicillin against S. aureus (0.98 µg/mL) and same potency as ciprofloxacin against S. pneumoniae (1.95 µg/mL). The g−ve activity of this compound 16b, was also demonstrated by its activity against S. typhimurium with the same potency as ciprofloxacin (3.9 µg/mL), 4-folds its activity against K. penumoniae and 2-folds the activity against E. coli. In particular, the arylidene-hydrazide derivatives 8b, d and g were the only derivatives in this series that showed activity against the g−ve resistant, Pseudomonas aeruginosa, where 8b and d were twice as potent as ciprofloxacin (1.95 and 3.9 µg/mL) and 8g was equipotent.
In vitro CytotoxicityThe cytotoxic activity of the most active antitubercular derivatives 8b, c and g were tested against normal breast cells WI-38 using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.37) The results are expressed as growth inhibitory concentration values (IC50) which represent the concentration of the compound needed to make a 50% inhibition of cell growth after 48 h of incubation compared to untreated control. None of the three compounds displayed significant cytotoxic effect (IC50=127.7, 98.2 and 146.04 µM, respectively), thereby providing a good therapeutic index (Fig. 2).
MATERIALS AND METHODS
General ChemistryMelting points were measured with a Stuart melting point apparatus and were uncorrected. IR spectra were recorded as potassium bromide discs on Shimadzu FT-IR 8400S spectrophotometer and expressed in wave number (cm−1). The NMR spectra were recorded by Bruker spectrophotometer at 400 MHz. 1H-NMR spectra were run at 400 MHz, while 13C-NMR spectra were run at 100 MHz in deuterated dimethylsulphoxide (DMSO-d6). Chemical shifts (δH) are reported relative to TMS as internal standard. All coupling constant (J) values are given in hertz. Chemical shifts (δC) are reported relative to DMSO-d6 as internal standards. The abbreviations used are as follows: s, singlet; d, doublet; m, multiplet. Mass spectra were measured on a GCMS-QP1000 EX and Helwett Packard 5988 spectrometers at 70 e.V. Elemental analyses was carried out at the Regional Center for Microbiology and Biotechnology, Al-Azhar University, Cairo, Egypt. Reaction courses and product mixtures were routinely monitored by TLC on silica gel precoated F254 Merck plates. Unless otherwise noted, all solvents and reagents were commercially available and used without further purification.
1-(4-Bromophenyl)-3-(dimethylamino)prop-2-en-1-one 3Compound 3 was prepared according to reported procedure38) (mp 79–80°C, reported 75–77°C38)).
Ethyl 6-(4-Bromophenyl)-2-methylnicotinate 5Compound 5 was prepared according to reported procedure39) (mp 74–75°C, reported 72–7440)).
6-(4-Bromophenyl)-2-methylnicotinohydrazide 6Compound 6 was prepared according to reported procedure30) (mp 239–241°C, as reported30)).
General Procedure for Preparation of N′-Arylidene-6-(4-bromophenyl)-2-methylnicotinohydrazides 8a–gThe appropriate aldehyde derivative 7a–g (1 mmol) was added to a solution of the 6-(4-bromophenyl)-2-methylnicotinohydrazide 6 (0.31 g, 1 mmol) in absolute ethyl alcohol (10 mL) and a catalytic amount of glacial acetic acid. The reaction mixture was heated under reflux for 3 h. The formed precipitate was filtered off while hot, washed with methanol, and finally crystallized from ethanol/dioxane to furnish the target compounds 8a–g with 65–83% yield.
6-(4-Bromophenyl)-N′-(2,4-dichlorobenzylidene)-2-methylnicotinohydrazide (8a)White crystals (yield 70%), mp 269–271°C.30)
6-(4-Bromophenyl)-N′-(2,6-dichlorobenzylidene)-2-methylnicotinohydrazide (8b)White crystals (yield 73%), mp 261–263°C; IR (KBr, ν cm−1): 3167 (NH) and 1651 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.53, 2.65 (2 s, 3H, CH3), 7.35–7.42 (m, 3H, Ar-H), 7.71 (d, 2H, Ar-H, J=8.40 Hz), 7.85–8.00 (m, 2H, Ar-H), 8.09 (d, 2H, Ar-H, J=8.48 Hz), 8.34, 8.62 (2 s, 1H, Ar-H), 11.96, 12.03 (2 s, 1H, NH, D2O exchangeable); Anal. Calcd for C20H14BrCl2N3O: C, 51.87; H, 3.05; N, 9.07. Found: C, 51.54; H, 3.09; N, 9.15.
6-(4-Bromophenyl)-N′-(3,4-dimethoxybenzylidene)-2-methylnicotinohydrazide (8c)White crystals (yield 83%), mp 233–235°C; IR (KBr, ν cm−1): 3150 (NH) and 1651 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.53, 2.65 (2 s, 3H, CH3), 3.58, 3.72, 3.81 (3 s, 6H, 2-OCH3), 6.92–7.20 (m, 2H, Ar-H), 7.37 (s, 1H, Ar-H), 7.70 (d, 2H, Ar-H, J=8.44 Hz), 7.85–8.00 (m, 2H, Ar-H), 8.09 (d, 2H, Ar-H, J=8.52 Hz), 8.24 (s, 1H, Ar-H), 11.80, 11.91 (2 s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ ppm: 23.43 (CH3), 55.52, 55.92 (OCH3), 55.98, 56.04 (OCH3), 108.73, 109.28, 111.93, 112.10, 117.06, 117.58, 120.89, 122.49, 123.41, 123.63, 127.25, 129.11, 129.24, 129.85, 132.24, 137.31, 137.51, 137.56, 137.79, 144.24, 148.58, 149.26, 149.55, 150.88, 151.35, 154.72, 155.16, 155.41, 156.40, 164.21, 170.21; Anal. Calcd for C22H20BrN3O3: C, 58.16; H, 4.44; N, 9.25. Found: C, 58.34; H, 4.48; N, 9.16.
6-(4-Bromophenyl)-N′-(2,5-dimethoxybenzylidene)-2-methylnicotinohydrazide (8d)White crystals (yield 80%), mp 235–237°C; IR (KBr, ν cm−1): 3170 (NH) and 1662 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.52, 2.66 (2 s, 3H, CH3), 3.51, 3.81 (2 s, 3H, OCH3), 3.77 (s, 3H, OCH3), 6.79–7.39 (m, 3H, Ar-H), 7.70 (d, 2 H, Ar-H, J=8.52 Hz), 7.83–8.00 (m, 2H, Ar-H), 8.10 (d, 2H, Ar-H, J=8.40 Hz), 8.37, 8.65 (2 s, 1H, Ar-H), 11.90, 11.96 (2 s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ ppm: 23.43, 23.50 (CH3), 55.46, 55.94 (OCH3), 56.64, 56.73 (OCH3), 109.55, 109.94, 113.73, 113.95, 117.01, 117.10, 117.54, 118.42, 123.11, 123.43, 123.65, 129.13, 129.27, 129.65, 130.60, 132.24, 137.39, 137.54, 139.83, 143.77, 152.51, 152.84, 153.42, 153.74, 154.72, 155.07, 155.46, 156.56, 164.23, 170.35; Anal. Calcd for C22H20BrN3O3: C, 58.16; H, 4.44; N, 9.25. Found: C, 57.82; H, 4.49; N, 9.18.
N′-(4-Bromo-2-hydroxybenzylidene)-6-(4-bromophenyl)-2-methylnicotinohydrazide (8e)White crystals (yield 70%), mp 241–242°C; IR (KBr, ν cm−1): 3182 (NH) and 1651 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.52, 2.67 (2 s, 3H, CH3), 6.78, 6.90 (2 d, 1H, Ar-H, J=8.68 & 8.76 Hz), 7.31, 7.42 (2 dd, 1H, Ar-H, J=8.68, 2.36 Hz), 7.70 (d, 2H, Ar-H, J=8.40 Hz), 7.82–7.96 (m, 2H, Ar-H), 8.01 (d, 1H, Ar-H, J=8.04 Hz), 8.09 (d, 2H, Ar-H, J=8.44 Hz), 8.32, 8.51 (2 s, 1H, Ar-H), 10.26, 11.14 (2 s, 1H, OH, D2O exchangeable), 12.09, 12.20 (2 s, 1H, NH, D2O exchangeable); Anal. Calcd for C20H15Br2N3O2: C, 49.11; H, 3.09; N, 8.59. Found: C, 48.83; H, 3.13; N, 8.68.
6-(4-Bromophenyl)-2-methyl-N′-(thiophen-2-ylmethylene)nicotinohydrazide (8f)White crystals (yield 78%), mp 245–246°C; IR (KBr, ν cm−1): 3170 (NH) and 1658 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.52, 2.65 (2 s, 3H, CH3), 7.04, 7.15 (2 t, 1H, Ar-H, J=4.40 & 4.76 Hz), 7.37, 7.48 (2 d, 1H, Ar-H, J=3.00 & 2.64 Hz), 7.69–7.72 (m, 3H, Ar-H), 7.81–7.98 (m, 2H, Ar-H), 8.09 (d, 2H, Ar-H, J=8.52 Hz), 8.28, 8.53 (2 s, 1H, Ar-H), 11.87, 11.95 (2 s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ ppm: 23.45, 23.55 (CH3), 117.12, 117.58, 123.46, 123.67, 128.31, 128.39, 129.00, 129.18, 129.26, 129.60, 129.70, 130.25, 130.93, 131.73, 132.25, 137.35, 137.53, 137.60, 137.69, 139.15, 139.33, 139.82, 143.48, 154.77, 155.15, 155.49, 156.47, 164.21, 170.08; Anal. Calcd for C18H14BrN3OS: C, 54.01; H, 3.53; N, 10.50. Found: C, C, 53.82; H, 3.56; N, 10.33.
6-(4-Bromophenyl)-2-methyl-N′-(pyridin-3-ylmethylene)nicotinohydrazide (8g)White crystals (yield 80%), mp 247–249°C; IR (KBr, ν cm−1): 3190 (NH) and 1662 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.53, 2.66 (2 s, 3H, CH3), 7.34, 7.48 (2 dd, 1H, Ar-H, J=7.76, 4.84 Hz), 7.70 (d, 2H, Ar-H, J=8.44 Hz), 7.85–8.01 (m, 2H, Ar-H), 8.10 (d, 2H, Ar-H, J=8.44 Hz), 8.15 (d, 1H, Ar-H, J=8.56 Hz), 8.38 (s, 1H, Ar-H), 8.62 (d, 1H, Ar-H, J=4.80 Hz), 8.88 (s, 1H, Ar-H), 12.09, 12.16 (2 s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ ppm: 23.42, 23.49 (CH3), 117.22, 117.58, 123.49, 123.70, 124.40, 124.49, 129.22, 129.27, 129.45, 130.23, 130.29, 130.52, 132.20, 132.25, 133.54, 134.03, 137.42, 137.51, 137.66, 142.09, 145.72, 148.78, 149.30, 150.97, 151.33, 154.86, 155.08, 155.58, 156.54, 164.50, 170.52; Anal. Calcd for C19H15BrN4O: C, 57.74; H, 3.83; N, 14.17. Found: C, 58.08; H, 3.87; N, 14.08.
5-Bromo-2-(2-(4-chlorophenyl)-2-oxoethoxy)benzaldehyde 11To a stirred mixture of 5-bromosalicylaldehyde 9 (0.2 g, 1 mmol) and anhydrous potassium carbonate (0.27 g, 2 mmol) in absolute ethyl alcohol, a solution of 2-bromo-1-(4-chlorophenyl)ethan-1-one 10 (0.23 g, 1 mmol) in absolute ethyl alcohol was added. The reaction mixture was stirred for 2 h at room temperature. The solid product was collected by filtration, washed with water and ether, and crystallized from isopropanol to afford white crystals of compound 11 (yield 55%), mp 165–166°C; IR (KBr, ν cm−1): 1670, 1715 (2C=O); 1H-NMR (DMSO-d6) δ ppm: 5.83 (2 s, 2H, –OCH2–), 7.65 (d, 2H, Ar-H, J=8.48 Hz), 7.76–7.79 (m, 2H, Ar-H), 8.02 (d, 2H, Ar-H, J=8.72 Hz), 8.07 (s, 1H, Ar-H), 10.41 (s, 1H, CHO); Anal. Calcd for C15H10BrClO3: C, 50.95; H, 2.85. Found: C, 51.27; H, 2.79.
N′-(5-Bromo-2-(2-(4-chlorophenyl)-2-oxoethoxy)benzylidene)-6-(4-bromophenyl)-2-methylnicotinohydrazide 12Compound 11 (0.35 g, 1 mmol) was added to a suspension of 6-(4-bromophenyl)-2-methylnicotinohydrazide 6 (0.31 g, 1 mmol) in absolute ethyl alcohol (10 mL) and a catalytic amount of glacial acetic acid. The reaction mixture was heated under reflux for 2 h. The solid formed was filtered, washed with hot ethanol then crystallized from ethanol/DMF to furnish the target compound 12 with (yield 64%), mp 236–238°C; IR (KBr, ν cm−1): 3155 (NH) and 1701 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.54, 2.67 (2 s, 3H, CH3), 5.72, 5.74 (2 s, 2H, –OCH2–), 7.02, 7.12 (2 d, 1H, Ar-H, J=8.92 & 8.96 Hz), 7.41, 7.53 (2 dd, 1H, Ar-H, J=8.92, 2.48 Hz), 7.53 (d, 2H, Ar-H, J=8.44 Hz), 7.70 (d, 2H, Ar-H, J=8.48 Hz), 7.88–8.04 (m, 5H, Ar-H), 8.10 (d, 2H, Ar-H, J=8.48 Hz), 8.48, 8.73 (2 s, 1H, Ar-H), 12.14 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C28H20Br2ClN3O3: C, 52.41; H, 3.14; N, 6.55. Found: C, 52.18; H, 3.17; N, 6.69.
4-(Morpholino/1-piperidino)benzaldehyde 15a, bCompound 15a, b was prepared according to reported procedure41) (mp 66–69°C, 61–64°C, respectively).
General Procedure for Preparation of the Target Compounds 16a, bThe appropriate aldehyde derivative 15a, b (1 mmol) was added to a solution of the 6-(4-bromophenyl)-2-methylnicotinohydrazide 6 (0.31 g, 1 mmol) in absolute ethyl alcohol (10 mL) and a catalytic amount of glacial acetic acid. The reaction mixture was heated under reflux for 4 h. The formed precipitate was filtered off while hot, washed with methanol, and finally crystallized from ethanol/dioxane to give the target compounds 16a, b.
6-(4-Bromophenyl)-2-methyl-N′-(4-morpholinobenzylidene)nicotinohydrazide (16a)White crystals (yield 75%), mp>300°C; IR (KBr, ν cm−1): 3185 (NH) and 1658 (C=O); 1H-NMR (DMSO-d6) δ ppm: 2.52, 2.65 (2 s, 3H, CH3), 3.11, 3.21 (2 t, 4H, morpholino-H, J=4.52 Hz), 3.67, 3.73 (2 t, 4H, morpholino-H, J=4.68 Hz), 6.87 (d, 1H, Ar-H, J=8.72 Hz), 7.00 (d, 1H, Ar-H, J=8.80 Hz), 7.26 (d, 0.5H, Ar-H, J=8.68 Hz), 7.58 (d, 1.5H, Ar-H, J=8.72 Hz), 7.70 (d, 2H, Ar-H, J=8.52 Hz), 7.81 (d, 0.5H, Ar-H, J=8.04 Hz), 7.90–7.99 (m, 2H, Ar-H), 8.09–8.13 (m, 2H, Ar-H), 8.19 (s, 0.5H, Ar-H), 11.69, 11.77 (2 s, 1H, NH, D2O exchangeable); Anal. Calcd for C24H23BrN4O2: C, 60.13; H, 4.84; N, 11.69. Found: C, 59.88; H, 4.89; N, 11.74.
6-(4-Bromophenyl)-2-methyl-N′-(4-(piperidin-1-yl)benzylidene) Nicotinohydrazide (16b)White crystals (yield 80%), mp 283–285°C; IR (KBr, ν cm−1): 3170 (NH) and 1661 (C=O); 1H-NMR (DMSO-d6) δ ppm: 1.53–1.59 (m, 6H, piperidino-H), 2.64 (s, 3H, CH3), 3.19 (t, 4H, piperidino-H), 6.84 (d, 1H, Ar-H, J=8.00 Hz), 6.97 (d, 1H, Ar-H, J=8.40 Hz), 7.21 (d, 0.5H, Ar-H, J=8.28 Hz), 7.54 (d, 1.5H, Ar-H, J=8.52 Hz), 7.71 (d, 2H, Ar-H, J=8.16 Hz), 7.81 (d, 0.5H, Ar-H, J=8.04 Hz), 7.90–7.97 (m, 2H, Ar-H), 8.09–8.16 (m, 2.5H, Ar-H), 11.64, 11.73 (2 s, 1H, NH, D2O exchangeable); Anal. Calcd for C25H25BrN4O: C, 62.90; H, 5.28; N, 11.74. Found: C, 62.69; H, 5.33; N, 11.81.
Single Crystal X-Ray CrystallographyThe characterization of compounds 8a and 11 were also conducted by a single crystal X-ray structural analysis. The structures were solved with direct method and refined by SHELXTL.42,43) The measurements were carried out on a Bruker SMART APEX IID8 Venture diffractometer. Crystallographic data of compounds 8a and 11 is deposited with the Cambridge Crystallographic Data Center with deposition number CCDC 1430062 and 1431357, respectively. The crystallographic structures, crystal packing, crystallographic data, refinement, selected geometric parameters and hydrogen-bond geometry of both compounds are illustrated in the Supplementary Materials.
Antimycobacterial ActivityM. tuberculosis (RCMB 010126) strain was provided from culture collection of the RCMB, Al-Azhar University, Cairo, Egypt. Isoniazide and pyrazinamide were used as reference drugs. Antimycobacterial activity of the synthesized compounds was evaluated using the microplate Alamar blue assay (MABA)36) which was performed in black, clear-bottomed, 96-well microplates (in order to minimize background effect). Outer perimeter wells were filled with sterile water to prevent dehydration in experimental wells. Initial compounds dilutions were prepared in dimethyl sulfoxide and subsequent twofold dilutions were performed in the microplates. 0.1 mL of 105 CFU/mL M. tuberculosis inoculum was added to wells, additional control wells consisted of bacteria only (B). Plates were incubated at 37°C. Starting at day 4 of incubation, 20 µL of alamarBlue solution (Alamar Biosciences/Accumed, Westlake, OH, U.S.A.) and 12.5 µL l of 20% Tween 80 were added added to the entire plate. Plates were then incubated at 37°C, and results were recorded at 24 h post-reagent addition at 590 nm. Percent inhibition was defined as: 1−(mean of test well/mean of B wells)×100. Visual MICs were defined as the lowest concentration of drug that prevented a color change.
Antimicrobial ActivityAll strains were provided from culture collection of the RCMB, Al-Azhar University, Cairo, Egypt. Antibacterial and antifungal activities were expressed as the diameter of inhibition zones; Agar well diffusion method was used as described earlier.44,45) Holes (1 cm diameter) were digger in the agar using sterile cork borer in sterile malt agar plates for fungi and sterile nutrient agar plates for bacteria, which had previously been uniformly seeded with tested microorganisms. The holes were filled by fungal filtrates (100 µL). Plates were left in a cooled incubator at 4°C for one hour for diffusion and then incubated at 37°C for tested bacteria and 28°C for tested fungi. Inhibition zones developed due to active antimicrobial metabolites were measured after 24 h of incubation for bacteria and 48 h of incubation for fungi. Amphotericin B and ciprofloxacin were used as antifungal and antibacterial positive control, respectively. The experiment was performed in triplicate and the average zone of inhibition was calculated.
MICMIC was performed by a serial dilution technique described by Irobi et al.,46) starting with 100-mmol concentration of all compounds dissolved in 1 mL DMSO and then reduced by successive twofold dilutions of stock solution using a calibrated micropipette. Amphotericin B and ciprofloxacin were used as the reference compounds for fungi and bacteria; respectively. The final solutions concentrations were 125, 62.50, 31.25, 15.63, 7.81, 3.90, 1.95, 0.98, 0.49, 0.24 and 0.12 µmol/mL. The microtiter plates were incubated at 37°C for tested bacteria and 28°C for tested fungi and were readied using microplate reader after 24 h for bacteria and after 48 h for fungi. In each case, triplicate tests were performed and the average was taken as final reading. MIC was expressed as the lowest concentration inhibiting test organism’s growth.
Cytotoxicity against WI-38WI-38 cells (normal breast cells), were obtained from American Type Culture Collection. The cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hyclone), 10 ug/mL of insulin (Sigma), and 1% penicillin–streptomycin. All of the other chemicals and reagents were from Sigma, or Invitrogen. Cytotoxicity was determined using MTT assay following a reported procedure.47) The IC50 was estimated from graphic plots of the dose response curve for each conc. using Graphpad Prism software (San Diego, CA, U.S.A.). The data presented are the mean of at least three separate experiments.