Regular Articles
Bavachin Induces Apoptosis through Mitochondrial Regulated ER Stress Pathway in HepG2 Cells
2018 年 41 巻 2 号 p. 198-207

詳細
2018 年 41 巻 2 号 p. 198-207
As a traditional herbal medicine, the fruits of Psoralea corylifolia L. (Fructus Psoraleae (FP)) have been widely used for the treatment of various skin diseases for hundred years. Recently, the emerging FP-induced toxic effects, especially hepatotoxicity, in clinic are getting the public’s attention. However, its exact toxic components and mechanisms underlying remain unclear. Bavachin, one of flavonoids in FP, has been documented as a hepatotoxic substance, and the present study aimed to determine the toxicity caused by bavachin and the possible toxic mechanisms involved using human hepatocellular carcinoma (HepG2) cells. Our results showed that bavachin could significantly inhibited cell proliferation and trigger the endoplasmic reticulum (ER) stress in a dose dependent manner. Downregulating ER stress using tauroursodeoxycholic acid (TUDCA) obvious attenuated bavachin-triggerd cell apoptosis. Then, small interfering RNA (siRNA) knock-down of Mitofusion2 (Mfn2) resulted in a remarkable aggravation of ER stress through the inhibition of the phosphorylation of protein kinase B (Akt). Additionally, suppression of reactive oxygen species (ROS) by ROS Scavenger (N-acetyl-l-cystein (NAC)) also reduced bavachin-induced ER stress. Taken together, our study demonstrated that bavachin-induced ER stress caused cell apoptosis by Mfn2-Akt pathway, and that ROS may participate upstream in this mechanism. Here, we not only provide a new understanding of ROS/Mfn2/Akt pathway in bavachin-induced cytotoxicity via the ER stress, but also identify a new specific intervention to prevent FP-induced hepatotoxicity in the future.
The dried matured fruits of Fructus Psoraleae (FP) have been widely used for the therapeutic effects on osteoporosis, osteomalacia, and skin diseases.1,2) In recent years, increasing clinical reports have indicated potential hepatotoxicity of FP in patients,3–5) for instance, Cheung et al. showed three cases of acute hepatitis after exposed to FP and its related medicine such as Bu-gu-zhi intramuscular injection,3) and concerns grow over the health effects of FP. However, the hepatotoxic components and the mechanism of FP-induced hepatotoxicity are not fully understood. Such grim situation paves the way for crucial research endeavors.
Bavachin (also named coryfolin), the main flavonoids in the fruit of FP, has multiple pharmacological activities including platelet aggregation,6) α-glucosidase activities7) and antibacterial.8,9) It has been ever reported that bavachin is a potential estrogen supplement for estrogen replacement therapy because of the osteoblastic activity.10,11) However, as a “double-edged sword,” bavachin could also cause obvious adverse effects. Both our early work and latest articles suggested that endoplasmic reticulum (ER) stress and related signal pathways might play an important role in bavachin-induced hepatotoxicity.
The ER is responsible for the maintenance of intracellular homeostasis by controlling protein synthesis, folding, modification and transport of Ca2+.12) And ER dysfunction will lead to the activation of unfolded protein response (UPR) due to accumulated misfolded proteins in ER, which is referred to as the ER stress.13) The ER stress includes three signaling branches, protein kinase R (PKR)-like ER kinase (PERK)-eukaryotic initiation factor-2α (eIF2α)-activating transcription factor 4 (ATF4), inositol-requiring enzyme 1 (IRE1)-X-box binding protein 1 (XBP1) and ATF6α. When the ER stress is triggered, glucose regulated protein 78 (GRP78), also knowns as BIP, the master of UPR, dissociated from three branches and activates the downstream molecules, activated ER stress sensors transport into the nucleus and will reestablish homeostasis via ER-associated protein degradation (ERAD) and the UPR.14)
Multiple studies have showed that abnormal ER stress in cells will activate apoptosis, which may cause serious liver injury in clinic. ER stress responses occur under various conditions such as inflammation, hypoxia or nutrient deprivation, will eventually result in different diseases in humans including genetic diseases, neurodegenerative diseases, metabolic diseases and cancer. For example, ER stress pathways contribute to the regulation of lipid metabolism. Both intrinsic and extrinsic stimuli-triggered ER stress in hepatocytes would cause lipid accumulation, cell death and the development of liver injury. Recently, ER stress has been a critical target for disease prevention and therapy, but the exact regulatory mechanisms involved in ER stress-induced hepatotoxicity are still unclear.
Perturbation of ER homeostasis leading to ER stress responses is regulated by various signals, such as mitochondrial pathway. Mitochondrial division and fusion events have been linked with ER stress responses, which ultimately lead to apoptosis in response to unresolved stress.15) This cross talk decides cells fate with respect to survival or death following ER stress. For instance, although the most typical Bcl-2 family directly leads to the intrinsic apoptosis pathway, these proteins also participate in regulation of ER functions and decide cell fate following ER stress.16) Therefore, mitochondrial plays crucial role in regulating cellular events including Ca2+ release and apoptosis. On the other hand, to achieve these functions, there is a dynamic network linking between ER and mitochondrial in response to cellular stimulation. The dynamic network is called mitochondrial-associated membranes (MAMs). Mitofusion2 (Mfn2) is a transmembrane guanosine triphosphatase (GTPase) located on the outer membrane of the mitochondrial and ER. It is a member of MAMs and functions as a key regulator in controlling mitochondrial fusion rate and establishing interactions between mitochondrial and ER.17,18) In Mfn2 ablation cells, distance between ER and mitochondrial increases which result in impairing of Ca2+ uptake. However, there is little information as to the pathways involved.
Reactive oxygen species (ROS) is another regulator produced from mitochondrial. Accumulation of ROS induces a number of toxic responses including DNA damage, ER stress and apoptotic cell death.19,20) There have been reported that ROS may be an upstream regulator in ER stress, but the involvement in bavachin-induced ER stress has not been studied yet.
In this study, we demonstrate that bavachin induces apoptosis mainly through ER stress in HepG2 cells. Mitochondrial related signals including ROS and Mfn2 participate in the bavachin-induced apoptosis pathway. We employed small-interfering RNA (siRNA) to ablate Mfn2, and evaluated the effect of Mfn2 on phosphor-protein kinase B (Akt) and ER stress. Our results suggest that Mfn2 critically mediates hepatocellular adaptation to ER stress through inhibiting phosphor-Akt, and ROS may act as an upstream regulator in this process. Targeting crosstalk between ER stress and mitochondrial in human hepatocellular carcinoma (HepG2) cells may provide a theoretical basis for the treatment and prevention of FP-induced hepatotoxicity in clinic.
Bavachin (E031951 purity>99%) was acquired from EFEBIO (Shanghai, China). Tunicamycin (TM) and N-acetyl-L-cysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, U.S.A.). Rabbit polyclonal antibodies against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (2118), CHOP (5554), ATF4 (11815), Akt (4685), phosphor-Akt (13038) and horseradish peroxidase conjugated secondary antibodies were obtained from Cell Signaling Technology (U.S.A.). BIP(GRP78) (ab21685) was from Abcam (U.S.A.). Dulbecco’s modified Eagle’s medium (DMEM) and phosphate buffered saline (PBS), were obtained from GIBCO BRL (Gainthersburg, MD, U.S.A.). Hoechst 33342, Mito Tracker®Red CMXROS-Special Packaging and Alexa Fluor®488 were obtained from ThermoFisher Scientific (U.S.A.). A dichlorodihydrofluorescence diacetate (DCFH-DA) kit was obtained from Beyotime (S0033). An Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit was purchased from Miltenyi (Miltenyi Biotec, Germany). Tauroursodeoxycholic acid (TUDCA) is purchased from EMD Millipore (Billerica, MA, U.S.A.).
HepG2 Cell Lines and CultureHepG2 cells (purchased from Peking Union Medical College), were maintained in monolayer culture at 37°C and 5% CO2 in DMEM (Invitrogen Gibco, Paisley, U.K.) supplemented with fetal bovine serum (FBS) (10%), penicillin (100 U·mL−1), streptomycin (100 U·mL−1).
Cell Proliferation Assay2×104 cells/well were seeded in 96-well plates and incubated for 24 h. The cells were treated with 0.1% dimethylsulfoxide (DMSO) (control group) and 0, 2.5, 5, 10, 20, 40, 80, 160 and 200 µmol/L bavachin for 24, 48 and 72 h. The cell free group was set as the blank control group, and four repeating pipettes were set in each group. After drug treatment, a cell viability assay kit CellTiter96®AQueous One Solution Reagent (MTS) was added to each well containing the culture medium. The plate was incubated at 37°C for 1–1.5 h in a humidified, 5% CO2 atmosphere. The absorbance of each well at 490 nm was recorded using a multilabel reader (Victor™ X5, Perkin Elmer, U.S.A.).
Annexin V-FITC and PI Staining AssayHepG2 cells were treated with 20 µmol/L bavachin for 12, 24 and 48 h. After the treatment, 1×106 cells were washed in 1 mL of 1×binding buffer and centrifuged at 300×g for 10 min. Supernatants were completely aspirated. Cell pellets were resuspended in 1×Binding Buffer, and 10 µL of Annexin V-FITC was added into each group with 1×106 cells. After incubating with Annexin for 15 min, 5 µL of PI solution was added immediately prior to analysis by flow cytometry.
High Content Screening AssayFor determination of mitochondrial integrity and ATF4 nuclear translation, 1×104 cells/well were seeded in 96-well assay plates that were black with a clear bottom and lid. After 24 h, the cells were treated with various concentrations of bavachin (0.1% DMSO) and the positive control (TM) for 3, 6, 12 and 24 h. 2× concentrated staining solution (maintenance medium containing 10 µM/L Hochest33342 and 1 µM/L Mito Tracker®Red CMXROS) was added at 50 µL/well for a total volume of 200 µL and the plate was incubated for 1 h at 37°C. Then, 100 µL of fixative solution (4% paraformaldehyde solution) was added into each well and incubated for 20 min at room temperature. Medium was removed and incubated with permeabilization (0.2% tritonX-100 in PBS) solution for 30 min at room temperature. Permeabilization solution was removed and cells were washed twice with PBS. Blocking buffer (200 µL) was added into each well and incubated for 60 min at room temperature. Then, 40 µL of appropriate primary antibody solution was added to each well and incubated for 120 min at room temperature. After removing primary antibody solution, the plate was rinsed three times with PBS and 50 µL of secondary antibody was added to each well before incubation for 60 min at room temperature while protected from light. The plate was then imaged and analyzed after rinsed three times with PBS. Cell images were acquired using the 4′-6-diamidino-2-phenylindole (DAPI) (Hoechst33342), FITC (Alexa Fluor 488 Phalloidin), and Cy3 filters using In Cell Analyzer 2000. Images were processed and quantified using the multiwavelength cell scoring application module in In Cell Analyzer 1000 Workstation 3.5 software.
Intracellular ROS Production AssayA DCFH-DA kit was used to inspect the generation of ROS in bavachin treated HepG2 cells. After incubation with various concentrations of bavachin, 10 µmol/L DCFH-DA was loaded on the cells and incubated for 30 min at 37°C in the dark. Then, the cells were harvested and washed with PBS. The fluorescence signal was measured and analyzed by flow cytometry.
Quantitative (q)RT-PCR AssayTotal RNA was extracted from HepG2 cells using Trizol reagent (Invitrogen), and reverse transcribed into cDNA using. qPCR was performed in a Real-Time PCR detection system (Gene Amp 2400), with SYBR Green Supermix (Bio-rad, U.S.A.) and relevant primers. The primers were as follows: ATF4 F, 5′-TGA AGG AGT TCG ACT TGG ATG CC-3′; ATF4 R, 5′CAG AAG GTC ATC TGG CAT GGT TTC-3′. ATF6 F, 5′-GGG AGT GAG CTG CAG GTG TA-3′; ATF6 R, 5′-TTA TGG GTG GTA GCT GGT AA-3′. GAPDH F, 5′-AAT CCC ATC ACC ATC TTC CAG-3′; GAPDH R, 5′-AGG GGC CAT CCA CAG TCT TCT-3′. The percentages of the above molecules were quantified using StepOne Plus™ Real Time PCR (Applied Biosystems, U.S.A., StepOne Plus™ 272006169).
Western Blot AssayHepG2 cells were treated with various concentrations of bavachin for the indicated times. At the end of treatment, the cells were harvested and sonicated in RIPA lysis buffer. Forty micrograms of proteins were resolved by 8–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinyliden fluoride (PVDF) membranes. The PVDF membranes with protein were blocked with blocking buffer (5% nonfat dry milk, in Tris-buffered saline containing 0.1% Tween-20; TBST) for 3 h, incubated with the primary antibody for 3 h at room temperature, which was followed by 4°C overnight. The membranes were incubated with secondary antibody for 1 h and visualized with a luminescent image analyzer (GE Healthcare Bio-Sciences AB, ImageQuant LAS 500, Sweden).
RNA Interference with Mfn2HepG2 cells were transfected with specific siRNA duplexes targeting Mfn2 in serum-free Opti-MEM (Gibco) using Lipofectamine2000 and incubated for 24 and/or 48 h. In this experiment, three different siRNAs were used to confirm that the inhibition of the pathway is from mRNA degradation instead of from siRNA addition itself. The transfection efficiency was confirmed by qRT-PCR and Western blot analysis. The siRNA used to target Mfn2 and a scrambled siRNA were synthesized by Ribo (China).
Statistical AnalysisIn this study, all results are from at least three independent experiments and data are expressed as the mean±standard deviation (S.D.). Data were analyzed by one-way ANOVA to test the significance of differences between the control and drug-treated groups. p<0.05 was considered statistically significant.
To investigate cell viability effects of bavachin in human hepatocytes, bavachin exerted an overt impact on the HepG2 cellular morphology from 6h treatment (Fig. 1A). HepG2 cells viability was detected using the MTS assay after exposed to various concentrations of bavachin for 24, 48 and 72 h. As shown in Fig. 1B, bavachin exhibited significant cytotoxic with an IC50 value of 24.4 µmol/L at 24 h. The flow cytometry results confirmed the effect of bavachin on cell fate over time (Fig. 1C). These results suggest that bavachin significantly suppresses the proliferation of HepG2 cells through activating cell apoptosis process in a dose-dependent and time-dependent manner. In order to determine the early molecular events, treatment with 5, 10, 20 µmol/L bavachin for 12–24 h were selected for the subsequent experiments according to the IC50 value.
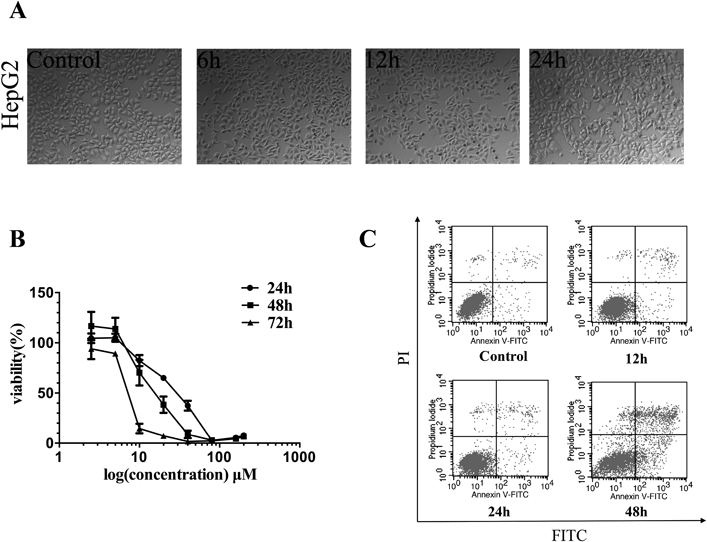
(A) Bavachin treatment alters cell morphology in HepG2. HepG2 cells were treated with 20 µmol/L bavachin for 6, 12, and 24 h, respectively, phase-contrast micrographs show cell morphology of the cells. (B) Bavachin inhibits HepG2 cells proliferation. HepG2 cells were treated with various concentrations of bavachin and estimated viability by the MTS assay. (C) Bavachin induces HepG2 cells apoptosis in a time-dependent manner. HepG2 cells were treated with 20 µmol/L bavachin for 12, 24, and 48 h. Cell apoptosis was estimated by Annexin V-PI kit. Values represent the mean±S.D. (n=3).
To test the possible involvement of ROS in bavachin-induced apoptosis, we examined whether bavachin affect ROS generation in HepG2 cells (Figs. 2A, B). Bavachin induced significantly increased ROS generation, which was evident at 12h after 40 µmol/L bavachin treatment. Simultaneously, bavachin treatment resulted in loss of mitochondrial membrane potential (MMP) (Figs. 2C, D). Furthermore, changes in mitochondrial morphology are clearly observed, including changes in the total area, width, height, length, and breadth, as shown in Figs. 2E, F. However, mitochondrial apoptosis related proteins including Bax and caspase-3 were not significantly affected by bavachin (Fig. 2G). These results indicate that bavachin indeed induced mitochondrial injury in HepG2 cells, while bavachin-induced apoptosis was not dependent on the classic mitochondria-mediated apoptosis (the caspase-dependent cell apoptosis).
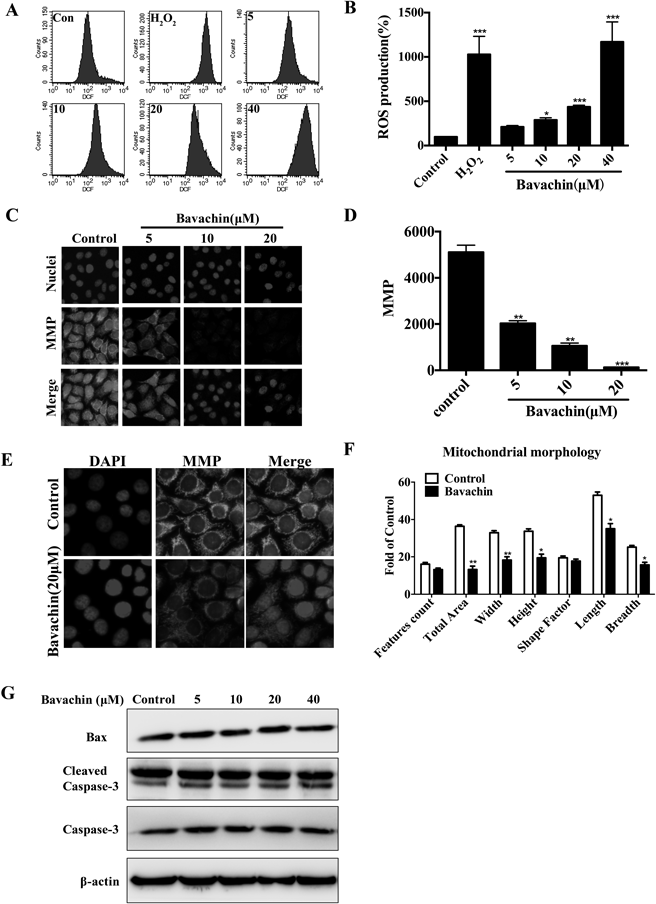
(A, B) Bavachin increases of generation of ROS. HepG2 cells were treated with various concentrations of bavachin for indicated time, the levels of ROS in cells were detected by DCFH-DA measurement using flow cytometry analysis. (C–F) Treatment with bavachin leads to mitochondrial dysfunction. HCS was used to present images and statistical analysis of MMP (C, D) and mitochondrial morphology changes (E, F). (G) Bavachin induces non-caspase dependent apoptosis. Unchanged expression of mitochondrial death signal proteins (Bax and cleaved caspase-3). The results are expressed as the mean±S.D.; * p<0.05 vs. control, ** p<0.01 vs. control, *** p<0.001 vs. control.
ER stress has been postulated as a common pathologic denominator in various metabolic diseases including drug-induced liver injury.21) Therefore, whether bavachin-induced ER stress would lead to activation of apoptosis need further investigation.
ER stress pathways consist of three sensor proteins including PERK, IRE1α and ATF6. When the ER stress is activated, BIP (GRP78), which is the master regulator of these sensor proteins, dissociated from PERK and phosphorylated eIF2α and then activates ATF4 translation. ATF4 increases the expression of pro-apoptotic factor CHOP. XBP1s is another ER stress response maker which is the downstream signal of IRE1α. Therefore, we first monitored the expression levels of ATF4 in bavachin treated HepG2 cells. We found that ATF4 nuclear intensity was elevated at 12h with 20 µmol/L bavachin treatment (Figs. 3A, B). We also found that bavachin activated CHOP and XBP1s in cells (Figs. 3C, D).
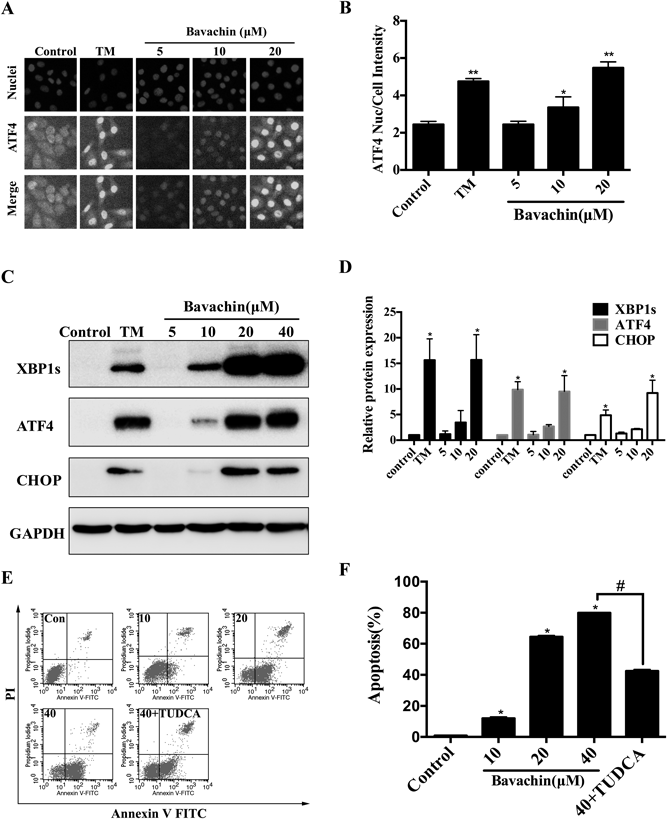
(A, B) Bavachin activates ATF4 nuclear translocation. HepG2 cells were treated with various concentrations of bavachin for 12 h, the levels of ATF4 nuclear intensity were analyzed by HCS. FITC stains shows increased intensity after 20 µmol/L bavachin treatment. (C, D) ER stress activation in HepG2 cells. Western blot analysis shows upregulation of ATF4, XBP1s and CHOP. (E, F) Inhibited ER stress prevents bavachin-induced apoptosis. HepG2 cells were pretreated with TUDCA (100 µmol/L) at 37°C for 30 min and then treated with bavachin for 12 h. Bavachin induced cell apoptosis were attenuated by ER stress inhibitor TUDCA. * p<0.05 vs. control, ** p<0.01 vs. control, # p<0.05 vs. bavachin (40 μM).
ER stress response dysfunction may initiate an apoptosis pathway.22) Treatment with bavachin caused apoptosis as evident by Annexin (Figs. 3E, F). TUDCA, an ER stress inhibitor, obviously suppressed ER stress and proportionally lowered bavachin-induced cell apoptosis. The percentage of apoptotic cells was decreased along with the inhibition ER stress responses, indicating a critical role of ER stress in the apoptotic cells.
Mfn2 Depletion Aggravated Bavachin-Induced ER StressIt has been reported that the outer mitochondrial membrane GTPase Mfn2 regulates ER shape in addition to its mitochondrial fusion effects.17,23) While, its role in bavachin-induced ER stress is unclear. To investigate the possible effect of Mfn2 in bavachin-induced ER stress and UPR, we first explored how bavachin (20 µmol/L) affect Mfn2 expression under bavachin treatment conditions. HepG2 cells exhibited gradual decreases in the Mfn2 protein levels, which were evident at 3 h after bavachin treatment and reached levels less than 30% of the controls by 9 h (Fig. 4A). Then, Mfn2 expression in cells was depleted using siRNA interference technology and we found that treatment with bavachin led to increased expression of ATF4 and XBP1s, and the induction of these proteins was generally higher in Mfn2-depletion HepG2 cells (Figs. 4B, C). Additionally, real-time qPCR showed significantly up-regulated mRNA levels of ATF4 and ATF6 for Mfn2-depletion HepG2 cells compared with the control HepG2 cells at baseline (Fig. 4D). Taken together, these results suggest that the Mfn2 depletion aggravated bavachin-induced ER stress, through both the three sensor pathways including PERK-ATF4, IRE1-XBP1s, and ATF6.
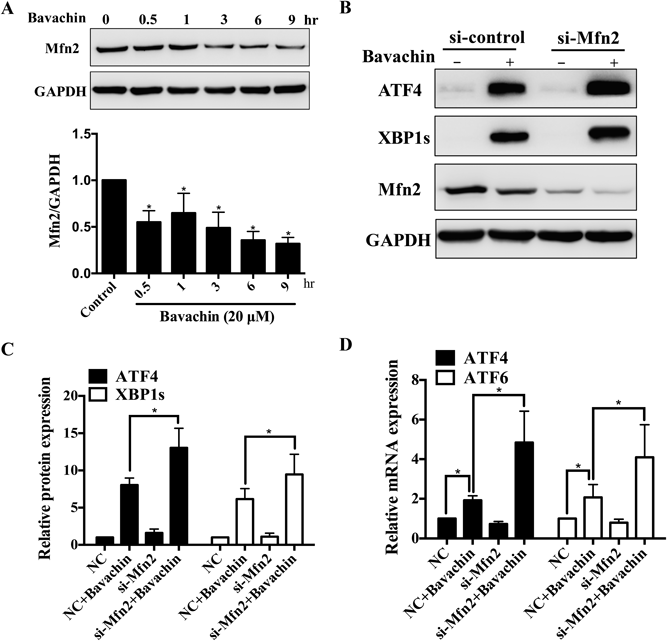
(A) Downregulation of Mfn2 in bavachin-treated HepG2 cells. Western blot analysis was performed 0–9 h after bavachin treatment of cells. Mfn2 expression levels were compared to corresponding GAPDH levels. (B, C) Mfn2 ablation aggravates ER stress. Mfn2 ablation by siRNA induces upregulation of ER stress response proteins in HepG2 cells. Western blot analysis was performed 48 h after the addition of control siRNA (NC), or Mfn2 siRNA (si-Mfn2) and 12 h with bavachin treatment. (D) Mfn2 ablation aggravates ER stress related molecular mRNA levels including ATF4 and ATF6. * p<0.05 vs. control.
After confirming the role of ER stress in bavachin-induced cell apoptosis, we next investigate the downstream signal of Mfn2-mediated ER stress. Akt plays a major role in regulating ER and mitochondrial apoptosis pathways.24,25) In this study, phosphor-Akt was significantly increased by bavachin in a time-dependent manner, while total Akt have no apparent change (Figs. 5A, B). In Mfn2 deficient HepG2 cells, increased phosphor-Akt was significantly suppressed in bavachin treated HepG2 cells, as shown in Figs. 5C and D. These results indicate that both Akt and Mfn2 participate in regulating bavachin-induced ER stress. Deficiency of Mfn2 aggravates ER stress through preventing increased phosphor-Akt. As a result, Mfn2 acted as an attenuator in response to ER stress via promoting phosphor-Akt.
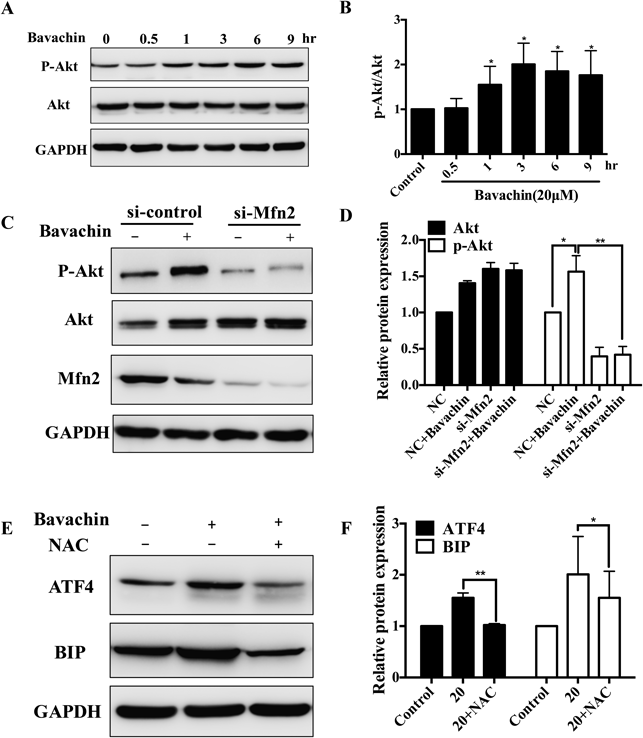
(A, B) Time dependent effect of bavachin on phosphor-Akt. Western blot analysis shows bavachin increased phosphor-Akt in a time-dependent manner. (C, D) Mfn2 siRNA inhibits phosphor-Akt in response to bavachin. Cells were transfected with control siRNA (NC) and Mfn2 siRNA (si-Mfn2) for 48 h and bavachin treatment for 12 h. Western blot analysis shows phosphor-Akt protein levels. The results are representative of three independent experiments. (E, F) NAC suppresses bavachin-induced ER stress. Cells were pretreated with 1 mM NAC for 1 h and then treated with bavachin for 12 h. ATF4 and BIP proteins levels were analyzed by Western blot assay. The results are expressed as the mean±S.D.; * p<0.05 vs. control, ** p<0.01 vs. control.
As mentioned above, bavachin induced mitochondrial injury including ROS production and loss of MMP, but did not activate mitochondrial apoptotic pathway. Whether ROS is the upstream regulator involving in bavachin-induced ER stress? Our data showed that pretreatment with 1 mM NAC for 1 h, suppressed the increased ROS and simultaneously completely inhibited activation of ATF4 and BIP induced by bavachin (Figs. 5E, F), suggesting that ROS plays an upstream role in bavachin-induced ER stress.
In this study, we have confirmed that bavachin inhibits HepG2 cells and induced apoptosis mainly through ER stress other than mitochondrial apoptosis. The data show that ROS accumulation, Mfn2 decrease and activated phosphor-Akt are responsible for bavachin-induced ER stress in HepG2 cells. ROS scavenger NAC significantly suppressed bavachin-induced ER stress, and results in restoration of MMP. While, mitochondrial fusion protein Mfn2 depletion could aggravate ER stress response by inhibiting phosphor-Akt. Thus, our study provides the first demonstration that bavachin-induced ER stress response is an inducible factor that lead to apoptosis in HepG2 cells. Moreover, we also proved that ROS and Mfn2 which are associated with mitochondrial function are central to the regulation of bavachin-induced ER stress.
Several studies have implicated the role of FP in apoptosis. Lv and Liu26) suggested the anti-proliferative role of bakuchiol in human gastric cancer cell. In addition, psoralen and isopsoralen induced tumor cell apoptosis or necrosis of osteosarcoma,27) and psoralen was associated with apoptosis in human cells.28) These results suggest that components in FP induced apoptosis may function as a factor of FP-induced liver injury. However, the mechanisms of bavachin toxicity in the cell death has not been well elucidated. This study demonstrated that bavachin inhibited HepG2 cell viability in a dose-dependent and time-dependent manner.
Evidences have shown that ER stress occurs in nearly all physiologies related to acute and chronic liver disease including alcoholic liver disease,29,30) nonalcoholic fatty liver disease,31) viral hepatitis,32,33) and drug-induced liver injury (DILI).34–36) The ER plays a crucial role in maintaining cellular homeostasis, regulating protein synthesis, Ca2+ transportation and drug metabolism.37) It is widely accepted that ER stress and the downstream UPR exert both protective and detrimental effects on the cell.38) Activated ER stress and the UPR could protect cell from cellular injury, but ER stress overload or block in the UPR activation in cells would result in cell apoptosis.39,40) ER stress can be triggered by various intrinsic or extrinsic factors including accumulation of misfolded proteins, inflammation, hypoxia or nutrient deprivation. And we performed high-content screening (HCS), a high-throughput imaging-based cellular analysis technology that enables the sensitive, detailed detection of multiple parameters simultaneously. It is widely used to provide data on prediction of hepatotoxicity including changes in the nucleus, mitochondrial function, cellular ROS generation and ER stress. We found that one of six active compounds in FP, bavachin, could induced obvious ER stress in ATF6 transfected HepG2 cells (Table S1), which suggested that ER stress might mediate FP-induced hepatotoxicity.
Besides, bavachin not only activated the UPR but also disrupted the MMP and triggered apoptosis in HepG2 cells. Mitochondrial dysfunction also leads to cell apoptosis through mitochondrial pathway which is characterized by its regulation of the Bcl-2 protein family.41) Mitochondrial dysfunction is induced by homodimerization of Bax or Bak within the outer mitochondrial membrane.42) In addition, egress of cytochrome c from mitochondrial intramembrane space into the cytosol promotes caspase-3 activation hepatocyte apoptosis.43) To further investigate that whether bavachin activate mitochondrial-apoptosis or ER stress-apoptosis pathway, mitochondrial-apoptosis proteins including caspase-3 and Bax were examined and there were no obvious changes in bavachin-treated cells. However, the apoptosis induced by bavachin was prevented by the most well-known chemical chaperones TUDCA. It is reported that TUDCA reduces ER stress through binding to the hydrophobic regions of proteins, and promoting the protein folding and aggregation.44,45) This result indicated that bavachin-induced apoptosis was associated with ER stress pathways.
To further explore the mechanisms which activate bavachin-induced ER stress, we examined the effect of Mfn2 deficiency on bavachin-induced ER stress. Mfn2 is a key protein which participates in mitochondrial fusion and transfer of Ca2+ from the ER to mitochondrial.46,47) Given the crucial role of ER and mitochondrial in cellular regulation, Mfn2 is a key bridge in cross talk between the two organelles. It has been reported that mouse embryonic fibroblasts with thapsigargin (TG) or TM treatment contribute to upregulation of Mfn2 protein levels, and loss of Mfn2 aggravates TM-induced ER stress.18) In hepatocellular carcinoma (HCC) studies, overexpression of the Mfn2 gene resulted in tumor cell apoptosis via mitochondrial pathways.48,49) Thus, Mfn2 may act as anti-apoptotic regulator during cell responses in vitro and in vivo.50) In particular, Mfn2 mutations are associated with a plenty of diseases, including cancer,49) obesity,17,51) diabetes52) and liver diseases. Sebastián et al.53) showed that liver specific Mfn2 knockout mice led to perturbation of metabolic homeostasis including intolerance and enhanced hepatic gluconeogenesis. Therefore, Mfn2 might be an important trigger target in bavachin-induced ER stress, which will lead to hepatocytes injury with prolonged activation.
However, the precise mechanism by which the Mfn2 trigger the downstream signals has not been well defined. As a matter of fact, in response to cellular stresses, Mfn2 are involved in the regulation of cell fate through a various of pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway, cytokine-cytokine receptor interaction, and focal adhesion.54–56) A recent study suggested that the overexpression of Mfn2 suppressed cancer progression through inhibition of Akt signaling.24) Furthermore, bavachin was reported to exhibit direct effect on Akt phosphorylation.11,57) In this study, we showed that Mfn2 ablation by the siRNA resulted in the inhibition of phosphor-Akt, and inhibition of phosphor-Akt also aggravated bavachin-induced ER stress (Fig. S1). Therefore, these studies imply that Mfn2 and phosphor-Akt might coordinately regulate translational responses of the cell to bavachin-induced ER stress. In tunicamycin treated HepG2 cells, we found the similar results (Fig. S2).
ROS are known mediators which are mainly produced by the mitochondrial respiratory chain and trigger a series of toxic events including apopotosis.58,59) Accumulation of ROS production will directly damage mitochondrial coupling with dysfunction of ER. In the present study, bavachin not only activated ER stress but also disrupted mitochondrial function evidenced by depletion of MMP and increased ROS. Interestingly, scavenging of ROS by antioxidant NAC successfully attenuated bavachin-induced ER stress and restored MMP at the same time. This result demonstrated that ROS was an initial signal leading to ER stress and mitochondrial injury.
Apoptosis is a regular physiological mechanism for eliminating cells in DILI. In addition to its importance as a biological phenomenon, excessive apoptosis leads to cell death with a various of diseases involved in it. Mitochondrial pathway and ER stress pathway are two major activation mechanisms of apoptosis. In this study, we found that ROS accumulation induced by bavachin decreased Mfn2 expression which further regulated ER stress through phosphor-Akt. Thus, Mfn2 may mediate the apoptosis from mitochondrial to ER. Taken together, these results showed that bavachin-induced apoptosis was associated with ER stress, and mitochondrial injury pathway (ROS/Mfn2/Akt) participated in this progress (Fig. S3).
In conclusion, we have defined bavachin induced ER stress related apoptosis in HepG2 cells. We postulate that the ER stress response may be involved in the apoptosis mechanism through ROS/Mfn2/Akt pathways in HepG2 cells. Further, more signals are needed to be investigated to clarify the upstream regulatory mechanisms of ER stress induced by bavachin, and we will continue to focus on the latest developments of bavachin toxic mechanisms in the future. The studies on ER stress and oxidative stress not only contribute to our understanding of the toxicity mechanisms of bavachin in FP, and would also provide guides for safety application of FP in clinic.
This work was supported by National Natural Science Foundation of China (No. 8163000131), the Beijing Natural Science Foundation (No. 7164291) and the Special Scientific Research Fund of State Administration of Traditional Chinese Medicine (2014ZX09304307-001-003, 2015ZX09501004-003-003, and 201507004); Department of Nephrology, Chinese People’s Liberation Army General Hospital, Chinese People’s Liberation Army Institute of Nephrology, State Key Laboratory of Kidney Disease, National Clinic Research Center for Kidney Diseases.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.