Abstract
Exosomes are tiny extracellular vesicles that are usually harvested in small quantities. Such small yield has been an obstacle for the expansion of the basic research regarding exosome analysis and applications in drug delivery. To increase exosome yield, we attempted to stimulate tumor cells via the addition of liposomes in vitro. Neutral, cationic-bare or PEGylated liposomes were incubated with four different tumor cell lines. The stimulatory effect of liposomal formulations on exosome secretion and cellular uptake propensity of the collected exosome by mother cells or different cells was evaluated. Both neutral and cationic-bare liposomes enhanced exosome secretion in a dose-dependent manner. Fluid cationic liposomes provided the strongest stimulation. Surprisingly, the PEGylation of bare liposomes diminished exosome secretion. Exosomes harvested in the presence of fluid cationic liposomes showed increased cellular uptake, but solid cationic liposomes did not. Our findings indicate that the physicochemical properties of liposomes determine whether they will act as a stimulant or as a depressant on exosome secretion from tumor cells. Liposomal stimulation may be a useful strategy to increase exosome yield, although further preparation to increase the purity of exosomes may be needed. In addition, fine-tuning of the biological properties of induced exosomes could be achieved via controlling the physicochemical properties of the stimulant liposomes.
Exosomes are extracellular nanoparticles (30–200 nm) secreted by most cells including normal and diseased cells such as tumor cells.1) They are present in biological fluids such as serum2) and urine.3) These natural nanoparticles have several functions in the biological milieu that may be beneficial or harmful. These functions include immune stimulation/tolerance, cell–cell communication, cellular resistance, and even tumor metastasis.2,4) Exosomes are also known to act as a defensive mechanism against any changes in the extracellular environment triggered by disease or stress factors.5)
Cell-derived exosomes are well recognized as efficient carriers of small RNAs to neighboring or distant cells, which has resulted in the preponderance of exosomes as carriers for gene therapy and other therapies over other artificial delivery carriers. Currently, much effort has been devoted to the development of exosome-based drug delivery systems for antioxidants, anticancer agents and antigenic peptides.6–8) However, the poor yield of exosomes from the supernatant of incubated cells poses a tremendous impediment to the progress of research on exosomes. Consequently, the development of an efficient isolation method as well as a resourceful induction method in order to collect large quantities of exosomes, which are secreted by incubated cells, is of utmost importance.
Exosome enrichment is a challenging task; many collection methods are currently available. The first accepted method was ultracentrifugation followed by purification with a sucrose gradient.9) Ultracentrifugation, however, has the disadvantage of being a time-consuming process that can lead to the degradation of biomolecules, which in turn, can result in a lowering of the purity of exosomes.10,11) Currently, many polymeric reagents can be used to isolate exosomes via precipitation, which is a process that is somewhat superior to ultracentrifugation in terms of purity and yield of exosomes.11) Nonetheless, almost all the currently applied methods failed to efficiently enrich the production level of exosomes. Accordingly, a novel approach to substantially augment the production of exosomes is urgently needed to ensure the widespread utilization of exosomes in basic research avenues including the drug delivery field.
Liposomes have been widely used as delivery carriers for anticancer agents and nucleic acids.12–14) Liposomes are known to interact with the cell surface in a physicochemical-dependent manner, which results in cell stimulation. Elsabahy and Wooley15) reported that nanomaterials could induce the production of cytokines in a variety of cells, particularly immune cells, and thus the level of cytokines could be used as a tool to evaluate the interactions between nanoparticles and cells, as in the process of immunotoxicity. In a similar manner, 1,2-dioleoyl-3-trimethylammonium propane chloride salt (DOTAP) cationic liposomes have been used to induce the expression of co-stimulatory CD80 and CD86 on dendritic cell surfaces, which started an immune response.16) Furthermore, lowering the positive charge on the surface of cationic nanoparticles via partial histamine modification has been used to diminish their immunotoxic response via a lowering of their interaction with cells.17) In addition, the lipid composition of liposomes frequently changes and their surface properties can be altered by many modification options such as PEGylation and the addition of cationic lipids. Changing the physicochemical properties of liposomes could impact exosome secretion from cells, and thereby, fine-tuning of the physicochemical properties of liposomes can be deployed as a viable means to enhance and/or attenuate the secretion of exosomes from cancer cells and consequently affect the yield of exosome collection.
In the present study, we investigated the response of tumor cells to different liposome preparations. It was found that stimulation with non-PEGylated bare liposomes increased exosome secretion from tumor cells in both a lipid-dose and a lipid-composition-dependent manner. Interestingly, PEGylation suppressed the secretion of exosomes from cancer cells.
MATERIALS AND METHODS
Materials and AntibodiesHydrogenated soy phosphatidylcholine (HSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), DOTAP, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethyleneglycol)-2000] (mPEG2000-DSPE) were generously donated by NOF (Tokyo, Japan). Cholesterol (CHOL) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). O,O′-Ditetradecanoyl-N-(α-trimethyl ammonio acetyl) diethanolamine chloride (DC-6-14) was purchased from Sogo Pharmaceutical (Tokyo, Japan). 3β-[N-(N′,N′-Dimethylaminoethane)-carbamoyl] cholesterol hydrochloride (DC-Chol) was purchased from Avanti Polar Lipids (LA, U.S.A.). Anti-TSG101 (ab30871) and horseraddish peroxidase (HRP) conjugated goat anti-rabbit immunoglobulin G (IgG) H&L (ab6721) were purchased from Abcam (Cambridge, U.K.). Anti-CD63 (sc-15363) antibody and anti-CD81 (sc-9158) were purchased from Santa Cruz Biotechnology (CA, U.S.A.). Exosome-depleted fetal bovine serum (exo-FBS) was purchased from System Biosciences (CA, U.S.A.). Normal FBS was purchased from Mediatech (CA, U.S.A.). All other reagents were of analytical grade.
Cell CultureFour cancer cell lines were purchased from the Cell Resource Center for Biomedical Research (RIKEN RBC CELL BANK, Saitama, Japan): the Colon 26 (C26) murine colorectal cancer cell line, the B16BL6 murine melanoma cell line, the MKN45 human gastric cancer cell line, and the DLD-1 human colorectal cancer cell line. They were employed as models for cancer cell lines in this study. They were maintained in RPMI1640 (Wako Pure Chemical Industries, Ltd.) supplemented with 10% exosome-depleted FBS (System Biosciences), 100 IU/mL penicillin, and 100 µg/mL streptomycin (MP Biomedicals, CA, U.S.A.) until reaching 80–90% confluency. All incubation processes were carried out under 5% CO2 at 37°C.
Preparation of LiposomesFour types of HSPC-based liposomes and six types of DOPE-based liposomes were prepared by the thin-film hydration method, as previously described.18) The lipid composition/molar ratio of the prepared liposomes is described in Table 1. In brief, the lipids were dissolved in chloroform and then lipid film was produced by removing the organic solvent via a rotary evaporator at 37°C under reduced pressure at 40 hPa for 1 h. The resultant lipid film was then hydrated using N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES) buffer (250 mM HEPES), 139 mM NaCl, adjusted to pH 7.4) at 65°C with shaking for 2 h. The resultant large multilamellar vesicles were then extruded through a polycarbonate membrane with pore sizes of 400, 200 and 100 nm using an extrusion device (Lipex Biomembranes Inc., VC, Canada). The diameters and zeta-potentials of prepared liposomes were determined in phosphate buffered saline (PBS) at 25°C using a Zetasizer Nano ZS (Malvern Instruments Ltd., WR, U.K.) (Table 1). Colorimetric assay was used to measure the phospholipid content of the prepared liposomes.19)
Table 1. Lipid Composition and Physicochemical Properties of Prepared Liposomes
| Composition (molar ratios) | Size (d.nm) | Zeta-potential (mV) |
|---|
| NL | HSPC/CHOL (2/1) | 130±1.72 | −0.95±0.62 |
| PEGylated NL | HSPC/CHOL/mPEG2000-DSPE (2/1/0.1) | 120±3.67 | −0.59±0.05 |
| CL1 | HSPC/CHOL/DC-6-14 (2/1/0.2) | 121±3.11 | +9.22±1.63 |
| PEGylated CL1 | HSPC/CHOL/DC-6-14/mPEG2000-DSPE (2/1/0.2/0.1) | 118±0.80 | −0.51±0.14 |
| CL2 | DOPE/DC-6-14 (2/1) | 137±2.45 | +22.20±1.49 |
| PEGylated CL2 | DOPE/DC-6-14/mPEG2000-DSPE (2/1/0.1) | 113±1.15 | +1.54±0.16 |
| CL3 | DOPE/DOTAP (2/1) | 154±9.56 | +21.37±1.45 |
| PEGylated CL3 | DOPE/DOTAP/mPEG2000-DSPE (2/1/0.1) | 98±0.18 | +0.36±1.01 |
| CL4 | DOPE/DC-Chol (2/1) | 177±3.74 | +17.5±0.42 |
| PEGylated CL4 | DOPE/DC-Chol/mPEG2000-DSPE (2/1/0.1) | 118±3.74 | +0.45±0.19 |
NL: Neutral bare liposomes, CL: Cationic bare liposomes.
Cancer cells were cultured in exosome-depleted conditioned medium for the indicated times, and then the cell culture medium was collected for exosomes (extracellular vesicle) enrichment. To collect exosomes (extracellular vesicle) secreted in response to liposome stimulation, the cancer cells were incubated for the indicated times in the presence of different liposome preparations of different lipid concentrations in exosome-depleted conditioned medium, and then the cell culture medium was collected for exosome (extracellular vesicle) isolation. Following collection of the culture medium, cells were harvested for cell viability determination using a Countess II automated cell counter (Thermo Fisher Science Inc., MA, U.S.A.) by staining cells with trypan blue.20) To remove cell debris in addition to the apoptotic bodies and microvesicles, the collected culture medium was exposed to differential centrifugations at 4°C (200×g for 10 min, 2000×g for 20 min and 12500×g for 30 min).9–11) Then, exosomes (extracellular vesicles) were enriched from the supernatant using either of the following two methods: ultracentrifugation (100000×g and 70 min) or an Exoquick-TC™ precipitation reagent (System Biosciences) according to the manufacturer’s recommended protocol.9–11,21) For the latter method, the reagent was added to the supernatant in a 1 : 5 ratio and then mixed well. The mixture was let stand at 4°C for 24 h. The supernatant was then completely discarded after two sequential centrifugation steps at 1500×g for 30 and 5 min. The exosome pellet was dispersed in PBS for further analysis and experiments. The liposomes in the incubation medium did not influence in the recovery ratio of exosomes under the experimental condition in this study. To confirm that the exosome samples contained no remains of the liposomes used in stimulation, exosome-depleted conditioned medium was incubated under similar experimental conditions in the presence of different liposome concentrations followed by precipitation using sequential centrifugations with ultracentrifugation or Exoquick-TC™. Then, the collected samples of liposome contaminants (without exosomes) were analyzed in the same manner as the exosome samples.
Characterization and Analysis of Collected Exosomes (Extracellular Vesicles)Bio-Rad DC® protein assay (Bio-Rad Laboratories Inc., CA, U.S.A.) was used to determine the protein concentration of the exosomes (extracellular vesicles) and liposome-bound proteins (liposome contaminants) according to the manufacturer’s recommended protocol. A linear standard curve with ovalbumin was used to calculate the protein concentration. To ensure a precise evaluation of exosome yield, the amount of proteins bound to the liposomes, which might have contaminated the collected exosome samples, was always subtracted from the final protein amount in the exosome (extracellular vesicle) sample. The amount of protein in the exosome (extracellular vesicle) samples was expressed as µg/106 viable cells or as µg/mL.
Exosome marker proteins (TSG101, CD63 or CD81) in the collected samples, containing exosomes and/or liposome-bound proteins) were identified by Western blot analysis. Briefly, protein samples were mixed with 2× sample buffer (0.1 M Tris, 4% sodium dodecyl sulfate (SDS), 12% 2-mercaptoethanol, 20% glycerol, slight amount of bromophenol blue) at a ratio of 1 : 1 (v/v), and then heated at 95°C for 5 min. Proteins in the samples were electrophoretically separated on 5–20% gradient gels (epagele-PAGEL; ATTO, Tokyo, Japan) at 25 mA per each gel for 70 min, as previously described.22) Each lane was loaded with 60 µg of protein. MagicMark™ XP Western Protein Standard (20–220 kDa, Thermo Fischer Inc.) was employed as a molecular weight standard. The separated proteins were blotted to a nitrocellulose membrane by electrophoresis at 12 V for 30 min using a semi-dry blotting system (ATTO). Then, for blocking, the membrane was incubated at 37°C for 1 h in the blocking buffer 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.05% Tween 20 (TBST 0.05%). The blocked membranes were further incubated with different primary antibodies in 2% BSA (in TBST 0.05%) in a 1 : 1000 (v/v) concentration for anti-TSG101 and a 1 : 40 (v/v) concentration for anti-CD63 and anti-CD81 at 4°C overnight. After that, the membranes were treated with HRP conjugated goat anti-rabbit IgG H&L antibody in TBST 0.05% with a dilution (1 : 20000) at 37°C for 1 h. Finally, membrane visualization was carried out by incubating the membrane with Amersham™ ECL™ Prime Western blotting Detection Reagent (Sigma-Aldrich, MO, U.S.A.) at room temperature for 5 min followed by imaging using image quant LAS 4000 (GE Healthcare Life Sciences, MA, U.S.A.).
Evaluation of the Cellular Uptake of Collected ExosomesTo assess the cellular uptake of collected liposomes, B16BL6 cells, which exhibited the highest exosomes yield among all tested cancer cell lines, was incubated for 48 h under both normal (exo-N) and stimulation conditions with 1 mM CL1 (exo-S1) or 0.05 mM CL3 (exo-S2). These two concentrations of CL1 and CL3 were selected as examples for the stimulating action of solid and fluid liposomes with a sublethal effect on cell viability. Exosomes were harvested from B16BL6 using ultracentrifugation at 100000×g for 70 min after the removal of cell debris, apoptotic bodies and microvesicles, as previously mentioned.9–11) The harvested exosomes from B16BL6 were evaluated for their cellular uptake by either the mother cell line B16BL6 or the allogeneic cell line C26. The exosomes were labeled using green fluorescent dye, PKH67 (Sigma-Aldrich), according to the manufacturer’s protocol with minor modifications.23–25) Briefly, a suspension containing the same amount of exosomes was washed once with PBS by ultracentrifugation at 100000×g for 70 min. The exosome pellets were re-suspended in diluent C supplied in the package (Sigma-Aldrich) and then mixed with an equal volume of the 2× dye solution in diluent C (2×10–6 M) for 5 min. The staining was stopped by the addition of an equal volume of exosome-depleted FBS. The stained exosomes were recovered as pellets by ultracentrifugation at 100000×g for 70 min. The pellets were then re-suspended in equal volumes of conditioned culture medium. Exosomes uptake was examined via flow cytometry (Gallios, Beckman Coulter, CA, U.S.A.) and confocal laser scanning microscopy (LSM 700, ZEISS) as described below. The liposome was incubated in exosome-depleted conditioned medium in the absence of cells. The collected supernatant was sequentially centrifuged as described above to obtain liposome contaminant that contained liposome-bound proteins. The liposome contaminants were stained and their cellular uptake was evaluated as described below.
Flow CytometryTarget cancer cells (B16BL6 or C26) were cultured at 1.5×105 cells in 2 mL of culture medium using a 6-well plate followed by incubation for 24 h. Then, labeled exosomes and/or liposome contaminants were incubated with cancer cells in exosome-depleted conditioned medium at a final protein concentration of 3 µg/mL of exosome sample or its equivalent of liposome contaminants. After 24 h post-incubation, the cancer cells were harvested, washed twice with PBS, and then examined by flow cytometry. The data were analyzed using Kaluza 1.2 software (Beckman Coulter).23,24)
Confocal Laser Scanning MicroscopyTarget cancer cells (B16BL6 or C26) were precultured for 24 h at a density of 3×104 cells in 200 µL of the exosome-depleted conditioned medium using Lab-Tek II chamber slides (Thermo Fischer Inc.). Labeled exosomes and/or liposome contaminants were added into each well at a final protein concentration of 3 µg/mL of exosome sample or its equivalent of liposome contaminants. The cells were then incubated for a further 24 h. After aspiration of the culture medium, adhered cells were washed with PBS and then incubated for 5 min in the presence of Hoechst 33342 DNA dye (1.78 µM) (Ana Spec Inc., CA, U.S.A.). After aspiration, cells were washed twice with PBS and then let stand for 30 min to dry. The dried cells were fixed with Fluoromount/Plus (Diagnostic Biosystems). Slides were examined at 63x magnification via confocal laser scanning microscopy. The scanned images were processed using LSM-ZEN2012 software (ZEISS).23–25)
Statistical AnalysisAll values were expressed as mean±standard deviation (S.D.) Statistical analysis was performed via one way ANOVA tests (Tukey’s and Dunnett’s multiple comparisons tests) using Graphpad Prism 6.01 software (GraphPad Software Inc., CA, U.S.A.). The level of significance was set at p<0.05.
RESULTS
Effect of Incubation Time and Type of Cancer Cell Line on Exosome (Extracellular Vesicle) SecretionTo trace the effect of incubation time on extracellular vesicle secretion from cancer cell lines, four different types of cancer cells were cultured for 24, 48 and 72 h, and the extracellular vesicles were then collected by Exoquick-TC™. Extracellular vesicle protein concentration was used as an indication of extracellular vesicle yield. Figure 1 shows that extracellular vesicle secretion was detected in all cell lines and that the extracellular vesicle yield was increased with incubation time in all cell lines except for DLD-1. Extracellular vesicle release after 72 h was in the following descending order: B16BL6 cells followed by DLD-1 then C26 and finally MKN45 by 786.72±69.92, 438.79±27.66, 393.05±33.24, and 376.10±72.96 µg/106 viable cells, respectively. These results manifest that all tested cancer cell lines can secret extracellular vesicles in both incubation time- and cancer cell-type-dependent manners with the highest level of extracellular vesicles secreted by B16BL6 cells following 72 h of incubation time.
Characterization of Exosomes (Extracellular Vesicles) Collected by Ultracentrifugation or Exoquick-TC™To characterize the samples obtained by either sequential centrifugations, the method widely used to obtain extracellular vesicles,9) or by Exoquick-TC™,21) protein levels were taken as an indicator for exosome yield. Proteins were obviously detected in the obtained extracellular vesicles regardless of the collection method (Fig. 2A). Exoquick-TC™ was likely to recover a large amount of extracellular vesicles compared with sequential centrifugations. Incubation of cells with neutral bare liposomes (NL) and cationic bare liposomes (CL) increased the protein amount in the collected extracellular vesicles regardless of the collection method (Fig. 2A). CL appeared to increase the production of extracellular vesicles much more than NL. It is well known that liposomes are easily interacted with serum proteins26,27) and the liposomes can then be precipitated by ultracentrifugation, which is similar to our experimental condition. Accordingly, we investigated the possibility that liposome-bound proteins were being contaminated in the collected extracellular vesicle fraction. As shown in Fig. 2B, the liposomes added into the exosome-depleted conditioned medium were precipitated together with serum proteins regardless of the collection method. To exclude the contribution of liposome-bound proteins to the overall protein concentration in the assayed sample, the amount of liposome-bound protein was subtracted from the overall protein concentration of the collected extracellular vesicles in all experiments conducted in this study. Figure 2B also showed that the contamination was higher with Exoquick-TC™ than ultracentrifugation. This is inconsistent with the literature which indicated that the protein contamination is higher with ultracentrifugation due to the degradation of large proteins by the high speed centrifugation and then precipitation with the collected exosome sample.10,11) Furthermore, to verify the existence of exosomes (extracellular vesicles) in the collected samples, the presence/absence of major exosome markers, such as (CD63, CD81 and TSG101),28,29) in the collected samples was scrutinized using the corresponding antibodies (Fig. 2C). All the examined markers were detected in all the collected samples incubated in the presence or absence of liposomes. However, such markers were not detected in the liposome-bound proteins fraction (data not shown). According to our current knowledge, the coexistence of exosomes and liposomes is novel to be investigated. Thus, other methods for exosome analysis like transmission electron microscopy (TEM), dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA) were unfortunately inconclusive to distinguish between exosomes and liposomes due to the similarity in their shape and size. Moreover, liposomes were already used as a model for exosomes in another study by Lane et al.30) Nevertheless, these results manifest that exosomes were efficiently obtained by either sequential centrifugations or Exoquick-TC™ and that the incubation of cells with liposomes (NL and CL) increased the secretion of exosomes. For the following study, Exoquick-TC™ was chosen to collect exosomes due to its efficiency in collecting larger amounts of them, which supported the detailed analysis of exosomes for this study.
Effect of Different HSPC-Based Liposome Preparations on Exosome SecretionTo further investigate the stimulatory effect of liposomes, the HSPC-based liposomes listed in Table 1 were incubated with different cancer cell lines for 48 h. As shown in Fig. 3, exosome secretions from all cell lines were increased with increasing phospholipid concentration (dose) of either neutral or cationic bare liposomes (NL or CL). Cationic bare liposomes (CL1) showed stronger stimulant activity on exosome secretion than neutral bare liposomes (NL) under the same experimental conditions, which is consistent with the results described earlier (Fig. 2). Among the tested cell lines, B16BL6 seems to be the most responsive cell line for liposomal stimulation in terms of exosome secretion followed by MKN45, DLD-1 and finally C26. Surprisingly, neither PEGylated NL nor PEGylated CL1 showed any stimulatory effect on exosome secretion, but these inhibited essential exosome secretion in a dose-dependent manner in some cell lines. This tendency was confirmed by the ultracentrifugation method (Supplementary Fig. S1). Taken together, these results show that bare liposomes have the ability to induce exosome secretion and that a cationic surface charge further increases the stimulatory effect of bare liposomes. On the other hand, it is likely that the PEGylation of these bare liposomes diminishes their stimulatory effect.
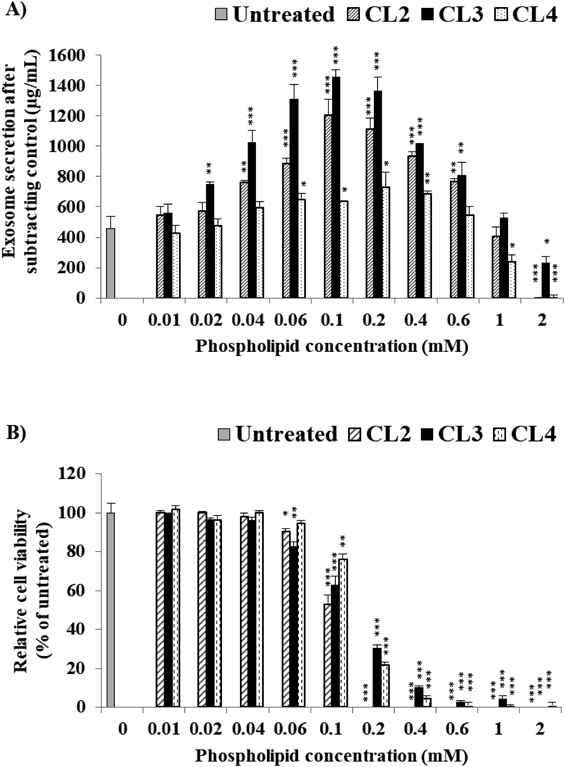
Effect of Cationic Lipid Type in Cationic Liposomes (CL) on Exosome SecretionTo gain further insight into the effect of the type of cationic lipids in the CLs of exosome secretion, C26 cells were selected due to having the lowest exosome yield, which required greater stimulation via powerful cationic lipid. C26 cells were incubated for 48 h in the presence of various CL preparations (CL2, CL3 and CL4), as listed in Table 1. All the tested DOPE-based CLs caused bell-shaped stimulation on exosome secretion in response to liposomal dose (Fig. 4A). DOPE-based cationic liposomes, tested at just 0.06 mM, showed higher stimulation activity on C26 (1.55-, 2.49- and 1.34-fold increases for CL2, CL3 and CL4, respectively) (Supplementary Fig. S2) compared with HSPC-based cationic liposome (0.5 mM, CL1) (Fig. 3A). Such stimulatory effect of DOPE-based cationic liposomes was mediated in a liposomal dose-dependent manner. At a lower liposomal dose, DOPE-based cationic liposomes could efficiently trigger exosomes secretion without significantly affecting cell viability (Fig. 4). On the other hand, at a higher liposomal dose, such stimulatory effect was substantially decreased (Fig. 4A) presumably via decreasing cell viability (Fig. 4B). The highest increase in exosome yield harvested at 0.1 mM was from CL3 followed by CL2 with 3.17- and 2.63-fold increases, respectively, compared with untreated cells. CL4 produced the lowest increase in exosome secretion (only 1.39-fold), which could have been due to a lowered level of membrane fluidity caused by an increase in the cholesterol content from DC-Chol in the liposomal membrane. Stimulation/inhibition observed in HSPC-based liposomes was also observed in DOPE-based liposomes (Supplementary Fig. S3). The dose-dependent stimulatory activity of cationic DOPE-based liposomes was inhibited by PEGylation as shown in Supplementary Fig. S3A (Exoquick-TC™) and Supplementary Fig. S3B (ultracentrifugation method). These results show that the stimulation of exosome secretion is affected by liposome lipid composition especially cationic lipid types in tandem with phospholipid types and cholesterol content.
Cellular Uptake of Harvested ExosomesThe role of exosomes in cell-cell communication is very important, particularly in the disease status of cells. Thus, exosome-based drug delivery is currently the focus of many studies, particularly in cancer therapy.6–8) In the present study, the cellular uptake of exosomes collected was studied either under normal conditions (exo-N) or under stimulation with 1 mM CL1 (exo-S1) and 0.05 mM CL3 (exo-S2). In addition, the cross-reactivity of exosomes harvested from a mother tumor cell line (B16BL6) towards another tumor cell line (C26) was investigated.
Control exosomes (exo-N) were taken up by the mother cells (B16BL6) as well as by other cells (C26) (Figs. 5A, 5B). The exo-S2 harvested by CL3 was also taken up by both cell lines (B16BL6 and C26) (Figs. 5A, 5B). Interestingly, there was little uptake of exo-S1 by the cancer cells (Figs. 5A, 5B). Compared with exo-N, exo-S2 was taken up by a higher percentage in both cancer cells (Fig. 5B). Notably, the uptake level of both exo-N and exo-S2 by mother cells (B16BL6) was higher than that by the C26 cells. Protein-bound liposome prepared with exosome-depleted conditioned medium showed very weak uptake signals with flow cytometry (negligible). The value was subtracted from the corresponding exosome sample. These observations were further emphasized when the exosomes inside target cells were visualized under LSM (Fig. 6). LSM images showed that green-labeled exo-N and exo-S2 were significantly internalized by the mother cells (B16BL6) as well as by other cancer cells (C26), while little faint green aggregations were detected in the case of exo-S1 (Fig. 6). The uptake of liposome associated with serum proteins from exosome-depleted conditioned medium was weak and negligible. It is likely that exosome uptake depends on the type of target cancer cell besides the type of liposomes used in stimulating exosome release.
DISCUSSION
Many studies have reported different stimulation models for exosome secretion from cells via manipulation of the cells, their receptors, the plasma membrane, or even intracellular electrolytes.31–33) These studies monitored only the stimulation action as a result of a specific condition or biological process. In the present study, it was demonstrated that in vitro incubation of bare liposomes with cancer cells enhances exosome secretion from different types of cancer cells, resulting in an increased yield of exosomes (Fig. 3). In particular, fluid cationic bare liposomes produced the greatest increase in exosome secretion at the optimum dose, which caused less cytotoxicity (Fig. 4). Thus, liposome-mediated stimulation of cancer cells could be a promising method to enrich exosome yield. Such an increased yield of exosomes supports the expansion of basic research regarding exosome analysis and their application as drug delivery vehicles, although further preparation to increase the purity of exosomes may be needed. Interestingly, PEGylation to the bare liposomes inhibited exosome secretion (Fig. 3 and Supplementary Fig. S3). The suppressive effect of PEGylation might be considered a new benefit for using PEGylated liposomes in treating tumors, because it has been reported that exosomes play a controversial role in tumor progress by stimulating/suppressing the immune system.2,4) Therefore, PEGylated liposomes may subside the bimodal role of exosomes, particularly in tumor metastasis. These observations suggest that our approach may be a new strategy to stimulate/inhibit the secretion of exosomes derived from cancer cells if the physicochemical properties of liposomes can be correctly manipulated.
The underlying mechanism behind increased exosome secretion via stimulation of liposomes remains uncertain. Raposo and Stoorvogel reported some different mechanisms for exosome release in a response to stimulation.33) For instance, the increased secretion of extracellular vesicles was triggered by stimulating p53 in tumor cells under stress conditions,34) via the activation of purinergic receptors, by changes in intracellular Ca2+ levels or by causing a depolarization of the cell membrane, which occurs by positively charged ions such as K+.31–33) The stimulatory effect of neutral bare liposomes (NL) and cationic bare liposomes (CL), observed in this study, might have been caused by these mechanisms. The study to reveal the mechanism behind the stimulatory effect is in progress in our laboratory.
The observed strong stimulation caused by CL rather than NL (Figs. 3, 4), is notably related to the cationic lipid in CL. Many studies have reported that CLs induce cytotoxicity in a dose-dependent manner.15,35) Therefore, the stimulatory effect of CL might reflect its cytotoxicity; more stress due to CL is applied to cancer cells, which may produce more exosomes as a defensive mechanism.5) The higher stimulatory effects of CL2 and CL3 are probably due to their higher interaction with tumor cells via not only surface cationic charge but also membrane fluidity,36,37) as well as to subsequent cell damage.35) It appears that membrane fluidity of liposomes also contributes to enhanced exosome secretion because both CL2 and CL3 produced exosomes at a higher rate compared with CL4, which contains cholesterol (DC-Chol) that creates a solid-phase membrane.
Interestingly, PEGylation to the surface of liposome suppressed secretion of exosome from the cells (Fig. 3 and Supplementary Fig. S3). The PEG conformation has a great effect on liposome-cell interactions; the mushroom structure of PEG on the surface of liposomes reduces nanoparticle–cell interactions, rather than the brush structure.38) In addition, the negative surface charge of PEGylated liposomes (Table 1) reduces or prevents the interaction between liposomes and cells due to electronic repulsion between these nanoparticles and negatively charged cell membranes.38) However, these factors cannot account for the suppression of the exosome secretion by PEGylation that was observed in the present study. Uz et al. recently found that the PEGylation of gold nanoparticles altered the cell cycle and caused DNA damage without apoptosis, which effectively disrupted cell division and replication. They showed that the effect was dependent on PEG grafting density and concentration; at a particular PEG grafting density (ca. 0.65 chains/nm2), none of these severe damages were observed.39) Literature studies have documented how the nanoparticles, which show no toxic effects according to classic toxicity test results, may severely disrupt cell-cycle steps, and cause DNA damage or apoptosis. Accordingly, the PEGylated liposomes in this study might have stimulated cells via liposome-cell collisions, which could have resulted in a suppression of exosome secretion. The mechanism behind this reaction will require further study.
The uptake of exosomes depends on many factors such as innate uptake ability of target cancer cell, characters of collected exosomes and the interaction between exosome and target cancer including adhesion, fitting surface antigen and fusion. Many studies have already reported that any change in exosome characters significantly affect their cellular uptake,40–42) which is consistent with our current observation (Fig. 5). The cellular uptake of exosomes obtained under normal and stimulated conditions extensively differed in a response to liposome type used in stimulation. The highest percentage of exosomes internalization was observed for exo-S2 followed by exo-N, while exo-S1 showed no detectable cellular uptake (Fig. 5). This might have been related to changes in the surface proteins of exosomes,43,44) which are responsible for cellular targeting and uptake. The similarities of the exosomal surface proteins with mother cells may explain the higher ability of B16BL6 to take up exosomes compared with the C26 cancer cell line. Further analysis of proteins in the collected exosome fraction is ongoing in our laboratory.
Liposomes have been widely used as carriers for chemotherapeutic agents and nucleic acids.12–14) Doxil®, doxorubicin-containing PEGylated liposome, has been approved for clinical use.45) Recently, many studies have indicated that exosomes have specialized functions and play a key role in processes such as intercellular signaling and waste management.2) Consequently, there is growing interest in the clinical applications of exosomes. In the present study, it was shown that liposomes have the ability to upregulate and/or downregulate exosome secretion in response to the surface modification of liposomes. After intravenous injection of long-circulating PEGylated liposomes, the liposomes reach solid tumors via an enhanced permeability and retention effect46) and might stimulate the tumor cells, resulting in a decrease in tumor-related exosome secretion in vivo. Exosomes are known to partially contribute to tumor metastasis.2,4) Therefore, chemotherapeutic agents containing PEGylated liposomes may provide a synergistic effect to tumor growth suppression as well as to the prevention of tumor metastasis.
In conclusion, our results show that in vitro incubation with liposomes enhances/suppresses exosome secretion derived from cancer cells. The stimulatory/inhibitory effect of liposomes is dependent on their dose, surface charge, membrane fluidity, and PEG modification, as well as on the type and viability of treated cancer cells. Liposomal stimulation may be a useful strategy to increase exosome yield, although further preparation to increase the purity of exosomes may be needed. In addition, our approach may be a new strategy to stimulate/inhibit the secretion of exosomes if the physicochemical properties of liposomes can be correctly controlled.
Acknowledgments
The authors are grateful to Mr. James L. McDonald for his helpful advice in developing the English manuscript. This study was supported by the Egyptian Government represented in the cultural affairs and missions sector of the Egyptian Ministry of Higher Education, and by a research program for development of intelligent Tokushima artificial exosome (iTEX) from Tokushima University, Takeda Science Foundation, the Uehara Memorial Foundation and Takahashi Industrial and Economic Research Foundation.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Hannafon BN, Ding WQ. Intercellular communication by exosome-derived microRNAs in cancer. Int. J. Mol. Sci., 14, 14240–14269 (2013).
- 2) Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol., 2, 569–579 (2002).
- 3) RamachandraRao SP, Matthias MA, Kokoy-Mondragon C, Aghania E, Park C, Kong C, Ishaya M, Madrigal A, Horng J, Khoshaba R, Bounkhoun A, Basilico F, De Palma A, Agresta AM, Awdishu L, Naviaux RK, Vinetz JM, Mauri P. Proteomic analysis of urine exosomes reveals renal tubule response to leptospiral colonization in experimentally infected rats. PLOS Negl. Trop. Dis., 9, e0003640 (2015).
- 4) Hedlund M, Nagaeva O, Kargl D, Baranov V, Mincheva-Nilsson L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE, 6, e16899 (2011).
- 5) Zhou Y, Xu H, Xu W, Wang B, Wu H, Tao Y, Zhang B, Wang M, Mao F, Yan Y, Gao S, Gu H, Zhu W, Qian H. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res. Ther., 4, 34 (2013).
- 6) Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles-endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta, 1846, 75–87 (2014).
- 7) Tan A, Rajadas J, Seifalian AM. Exosomes as nano-theranostic delivery platforms for gene therapy. Adv. Drug Deliv. Rev., 65, 357–367 (2013).
- 8) Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. BioEssays, 33, 737–741 (2011).
- 9) Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol., 3, 22–29 (2006).
- 10) Zlotogorski-Hurvitz A, Dayan D, Chaushu G, Korvala J, Salo T, Sormunen R, Vered M. Human saliva-derived exosomes: comparing methods of isolation. J. Histochem. Cytochem., 63, 181–189 (2015).
- 11) Van Deun J, Mestdagh P, Sormunen R, Cocquyt V, Vermaelen K, Vandesompele J, Bracke M, De Wever O, Hendrix A. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles, 3, 24858 (2014).
- 12) Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov., 13, 813–827 (2014).
- 13) Zhang Y, Satterlee A, Huang L. In vivo gene delivery by nonviral vectors: overcoming hurdles? Mol. Ther., 20, 1298–1304 (2012).
- 14) Huang SL. Liposomes in ultrasonic drug and gene delivery. Adv. Drug Deliv. Rev., 60, 1167–1176 (2008).
- 15) Elsabahy M, Wooley KL. Cytokines as biomarkers of nanoparticle immunotoxicity. Chem. Soc. Rev., 42, 5552–5576 (2013).
- 16) Cui Z, Han SJ, Vangasseri DP, Huang L. Immunostimulation mechanism of LPD nanoparticle as a vaccine carrier. Mol. Pharm., 2, 22–28 (2005).
- 17) Shrestha R, Elsabahy M, Florez-Malaver S, Samarajeewa S, Wooley KL. Endosomal escape and siRNA delivery with cationic shell crosslinked knedel-like nanoparticles with tunable buffering capacities. Biomaterials, 33, 8557–8568 (2012).
- 18) Ishida T, Harada M, Wang XY, Ichihara M, Irimura K, Kiwada H. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control. Release, 105, 305–317 (2005).
- 19) Bartlett GR. Colorimetric assay methods for free and phosphorylated glyceric acids. J. Biol. Chem., 234, 469–471 (1959).
- 20) Yao Y, Wei W, Sun J, Chen L, Deng X, Ma L, Hao S. Proteomic analysis of exosomes derived from human lymphoma cells. Eur. J. Med. Res., 20, 8 (2015).
- 21) Yim N, Ryu SW, Choi K, Lee KR, Lee S, Choi H, Kim J, Shaker MR, Sun W, Park JH, Kim D, Heo WD, Choi C. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun., 7, 12277 (2016).
- 22) Kawanishi M, Hashimoto Y, Shimizu T, Sagawa I, Ishida T, Kiwada H. Comprehensive analysis of PEGylated liposome-associated proteins relating to the accelerated blood clearance phenomenon by combination with shotgun analysis and conventional methods. Biotechnol. Appl. Biochem., 62, 547–555 (2015).
- 23) Ekström K, Valadi H, Sjöstrand M, Malmhäll C, Bossios A, Eldh M, Lötvall J. Characterization of mRNA and microRNA in human mast cell-derived exosomes and their transfer to other mast cells and blood CD34 progenitor cells. J. Extracell. Vesicles, 1, 18389 (2012).
- 24) Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z, Watkins SC, Falo LD Jr, Thomson AW. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood, 104, 3257–3266 (2004).
- 25) Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, Coscia C, Iessi E, Logozzi M, Molinari A, Colone M, Tatti M, Sargiacomo M, Fais S. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem., 284, 34211–34222 (2009).
- 26) Ishida T, Harashima H, Kiwada H. Liposome clearance. Biosci. Rep., 22, 197–224 (2002).
- 27) Ishida T, Harashima H, Kiwada H. Interactions of liposomes with cells in vitro and in vivo: opsonins and receptors. Curr. Drug Metab., 2, 397–409 (2001).
- 28) Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet., 21 (R1), R125–R134 (2012).
- 29) Carrière J, Barnich N, Nguyen HT. Exosomes: from functions in host-pathogen interactions and immunity to diagnostic and therapeutic opportunities. Rev. Physiol. Biochem. Pharmacol., 172, 39–75 (2016).
- 30) Lane RE, Korbie D, Anderson W, Vaidyanathan R, Trau M. Analysis of exosome purification methods using a model liposome system and tunable-resistive pulse sensing. Sci. Rep., 5, 7639 (2015).
- 31) Savina A, Fader CM, Damiani MT, Colombo MI. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic, 6, 131–143 (2005).
- 32) Lachenal G, Pernet-Gallay K, Chivet M, Hemming FJ, Belly A, Bodon G, Blot B, Haase G, Goldberg Y, Sadoul R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci., 46, 409–418 (2011).
- 33) Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol., 200, 373–383 (2013).
- 34) Lespagnol A, Duflaut D, Beekman C, Blanc L, Fiucci G, Marine JC, Vidal M, Amson R, Telerman A. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6/Steap3-null mice. Cell Death Differ., 15, 1723–1733 (2008).
- 35) Chou TH, Liang CH, Lee YC, Yeh LH. Effects of lipid composition on physicochemical characteristics and cytotoxicity of vesicles composed of cationic and anionic dialkyl lipids. Phys. Chem. Chem. Phys., 16, 1545–1553 (2014).
- 36) Kočišová E, Antalík A, Procházka M. Drop coating deposition Raman spectroscopy of liposomes: role of cholesterol. Chem. Phys. Lipids, 172-173, 1–5 (2013).
- 37) Socaciu C, Jessel R, Diehl HA. Competitive carotenoid and cholesterol incorporation into liposomes: effects on membrane phase transition, fluidity, polarity and anisotropy. Chem. Phys. Lipids, 106, 79–88 (2000).
- 38) Hu Y, Xie J, Tong YW, Wang CH. Effect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cells. J. Control. Release, 118, 7–17 (2007).
- 39) Uz M, Bulmus V, Alsoy Altinkaya S. The effect of PEG grafting density and hydrodynamic volume on gold nanoparticle-cell interactions: an investigation on cell cycle, apoptosis and DNA damage. Langmuir, 32, 5997–6009 (2016).
- 40) Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. U.S.A., 110, 17380–17385 (2013).
- 41) Inder KL, Ruelcke JE, Petelin L, Moon H, Choi E, Rae J, Blumenthal A, Hutmacher D, Saunders NA, Stow JL, Parton RG, Hill MM. Cavin-1/PTRF alters prostate cancer cell-derived extracellular vesicle content and internalization to attenuate extracellular vesicle-mediated osteoclastogenesis and osteoblast proliferation. J. Extracell. Vesicles, 3, 23784 (2014).
- 42) Smyth TJ, Redzic JS, Graner MW, Anchordoquy TJ. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta, 1838, 2954–2965 (2014).
- 43) Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles, 3, 24641 (2014).
- 44) McKelvey KJ, Powell KL, Ashton AW, Morris JM, McCracken SA. Exosomes: Mechanisms of Uptake. J. Circ. Biomark, 4, 7 (2015).
- 45) Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int. J. Nanomedicine, 7, 49–60 (2012).
- 46) Park JW. Liposome-based drug delivery in breast cancer treatment. Breast Cancer Res., 4, 95–99 (2002).