Abstract
Cells induce heat shock proteins (HSPs) against various stress. However, murine erythroleukemia (MEL) cells do not express HSP72, a heat-inducible member of HSP70 family. So, it is of interest to examine how MEL cells respond to heat stress (44°C, 30 min). Heat stress-induced apoptosis was suppressed by pretreatment of heat shock (44°C, 10 min). Such suppressive effects were maximal at 6 h after heat shock and remained up to 12 h. Interestingly, such effects of heat shock were abrogated by specific inhibitors of HSP90 such as 17-allylamino-17-demethoxygeldanamycin (17-AAG) and novobiocin. From flow cytometric analysis, it was found that MEL cells arrest in G2 phase at 6 h after heat shock, but restore original cell cycle at 12 h. High expression level of HSP90 was maintained before and after heat shock. Phosphorylation of HSP90α was observed in apoptotic cells induced by heat stress, but inhibited by pretreatment of heat shock. Such inhibition was abrogated by 17-AAG. Moreover, c-Jun NH2-terminal kinase (JNK) was activated in heat stress-induced apoptotic cells. Taken together, these results suggest that HSP90α in MEL cells plays an important role in the thermotolerance, i.e., suppression of heat stress-induced apoptosis by heat shock.
Mammalian cells respond through signal transduction pathway to physicochemical stresses such as UV irradiation,1) heating,2) and high pressure.3) The fate of these cells is determined depending on degrees and properties of cell damages. Particularly, the response of cells to heating is interesting from the use of hyperthermia to kill tumor cells.4) When mammalian cells are heated at 40–45°C for a short time, heat shock proteins (HSPs) such as HSP27, HSP72, and HSP90 are induced to protect the cells against elevated temperature.5–7) HSPs are molecular chaperones that protect cells from stressful conditions by preventing protein denaturation and repairing such damages.8,9) Of these HSPs, HSP72 is a heat-inducible member of HSP70 family and play an important role in the protection of heated cells.10) Interestingly, HSP72 is deficient in murine erythroleukemia (MEL) cells.11) Alternatively, HSP90 including major two isoforms, HSP90α and HSP90β, is relatively abundant and interacts with 200 client proteins which are associated with transcription factors and cell cycle.8) Particularly, the expression levels of intracellular HSP90 remain high in cancer cells.12)
If cells damaged by heating are unable to repair, heated cells undergo apoptosis.13) In apoptosis induced by heating, ceramide is generated through sphingomyelin hydrolysis by neutral shingomyelinase.13,14) Ceramide followed by ceramide metabolites such as sphingosin activates mitogen-activated protein kinases (MAPKs) and induces apoptosis and autophagy.15) As MAPK signal transduction of cell death, both signal pathways of p38 and c-Jun NH2-terminal kinases (JNKs) are well known in mammals.7,14) However, if the cells are pretreated under mild heating conditions, where apoptosis is not induced, such cells acquire the resistance against severe stress which induces apoptosis.10) For instance, heat stress (44°C, 30 min)-induced apoptosis in mouse embryofibroblasts (MEF) is suppressed by pretreatment at 44°C for 10 min (referred to as heat shock in the present paper).10) In the case of MEF, HSP72 that is induced by heat shock plays an important role in the suppression of heat stress-induced apoptosis.10) However, no HSP72 is expressed in MEL cells.11) So, it is of interest to examine whether heat stress-induced apoptosis in MEL cells is suppressed by pretreatment with heat shock. In the present work, we demonstrate that MEL cells acquire thermotolerance by heat shock so that become resistant to heat stress. In MEL cells, HSP90α activity is associated with such thermotolerance.
MATERIALS AND METHODS
Chemicals and MaterialsCompounds were obtained from the following sources: Acetyl-aspartic acid (Asp)-glutamic acid (Glu)-Val-Asp-4-methylcoumaryl-7-amide (Ac-DEVD-MCA), Peptide Institute Inc. (Osaka, Japan); 17-allylamino-17-demethoxygeldanamycin (17-AAG), ribonuclease A (RNase A) from bovine pancreas, Sigma (U.S.A.); Blocking One-P, Nonidet P-40 (NP-40), sodium fluoride, streptomycin sulfate, penicillin G potassium salt, phenylmethylsulfonyl fluoride (PMSF), propidium iodide (PI), Nacalai Tesque (Kyoto, Japan); novobiocin monosodium salt, Kanto Chemical Co. (Tokyo, Japan); sodium orthovanadate (V), Wako (Osaka, Japan). Rabbit polyclonal antibodies against the following targets were used: β-actin, Gene Tex. (U.S.A.); HSP72 (SPA-811), Stressgen (U.S.A.); HSP90 (4874), p-HSP90α-T5/7 (3488), Cell Signaling Technology (U.S.A.); p-JNK 1, 2, 3-T183, -Y 185 (bs-1640R), Bioss (U.S.A.); Serine/Threonine-protein kinase 38 (STK38, SAB1410182), Sigma-Aldrich (U.S.A.); survivin (sc-10811), Santa Cruz Biotechnology (U.S.A.). As a secondary antibody, horseradish peroxidase-conjugated goat antirabbit immunoglobulin G (12-346, Millipore, U.S.A.) was used. All other chemicals were of reagent grade.
Cell Culture and Heat TreatmentMEL cells (cell line 745 A) were maintained in the RPMI-1640 medium containing 10% fetal calf serum, streptomycin (0.1 mg/mL) and penicillin G (100 U/mL) at 37°C in a CO2 (5%) incubator. For the application of heat shock to MEL cells, the cells in the medium were exposed to 44°C for 10 min. These heat-shocked cells were suspended in fresh medium and cultured for 2–72 h at 37°C in the presence or absence of HSP90 inhibitors such as 17-AAG (1–5 µM) and 0.1 mM novobiocin. For heat stress, on the other hand, the MEL cell suspensions with or without HSP90 inhibitors were subjected to 44°C for 30 min. These heat-stressed cells were cultured at 37°C for 12 or 24 h in the presence or absence of HSP90 inhibitors. For proliferation ability of MEL cells, heat-shocked MEL cells were cultured for 0, 24, and 48 h. Then, portions of these cells were suspended in fresh medium and cultured during 24 h. The cell number was calculated by using a hemocytometer and an Olympus light microscope (model BHC, Japan).
ApoptosisFor detection of apoptotic cells, MEL cells in the medium were centrifuged for 5 min at 200×g and 4°C. The pellets were suspended in PBS (phosphate-buffered saline, 10 mM sodium phosphate, 150 mM NaCl, pH 7.4) and centrifuged under the same conditions. The MEL cells were fixed with 70% ethanol overnight at −20°C, and washed once with PBS. For the nuclear morphology, nuclei in the cells were stained with PI (50 µg/mL) and observed using a fluorescence microscope (Olympus, model IX-71). For the analysis of apoptosis using a flow cytometer (EPICS XL System II, Coulter, U.S.A.), the fixed cells in PBS were treated with RNase A (20 µg/mL) for 30 min at 37°C, washed once with PBS, and stained with PI (50 µg/mL).
Caspases are activated in apoptotic cells. So, the MEL cells were treated with aqueous solution containing 1% Triton X-100 and 1% NP-40. The lysate was incubated for 30 min at 4°C, and centrifuged for 5 min at 17000×g and 4°C. The caspase activity of supernatants was measured using an Ac-DEVD-MCA as substrate, as described previously.16)
Western BlottingMEL cells were incubated for 30 min at 0°C in a lysis buffer containing 1% Triton X-100, 1% NP-40, 0.5 mM PMSF, 1 mM sodium orthovanadate, and 5 mM sodium fluoride. The lysates were centrifuged at 18000×g for 5 min at 4°C and the resulting supernatants were used as cell extracts. Proteins in extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 8.5% acrylamide gel, according to the method of Laemmli.17) Proteins on gel were transferred onto polyvinylidene fluoride (PVDF) membranes. As the blocking solution, Blocking One-P was used. The transferred proteins were detected using primary antibodies, secondary antibody, and enhanced chemiluminescence detection kit (WSE-7120 L, ATTO, Japan). Densitomeric analysis of immunoblots was performed using an ATTO imaging system (Ez-Capture MG).
RESULTS
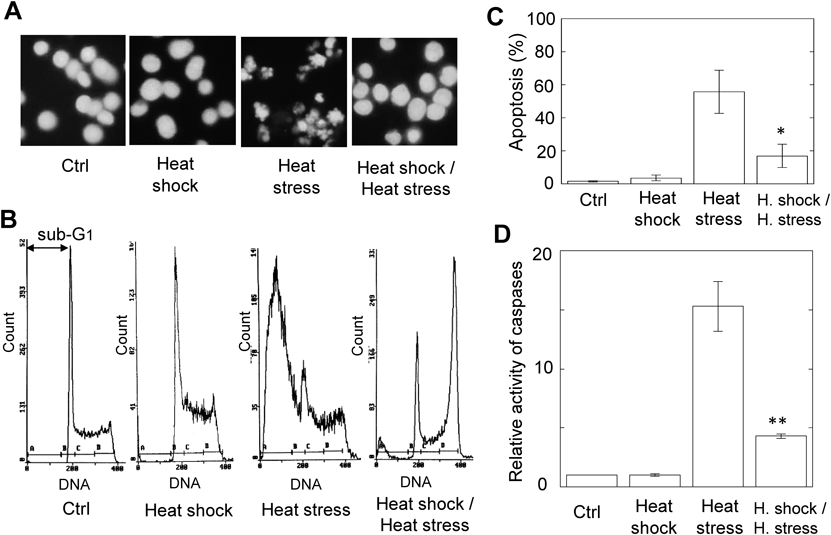
Heat Stress-Induced Apoptosis Is Suppressed by Heat Shock PretreatmentWhen the MEL cells exposed to the heat shock were cultured for 12 h at 37°C, the nuclear morphology in these cells was similar to that in intact cells (Ctrl) (Fig. 1A). On the other hand, the nuclear fragmentation, which is characteristic of apoptotic cells, was observed in the MEL cells subjected to the heat stress (Fig. 1A). Interestingly, such a fragmentation of nuclei was greatly suppressed when the MEL cells subjected to the heat shock were cultured for 6 h at 37°C before the heat stress (Fig. 1A). To estimate quantitatively the effect of heat shock on heat stress-induced apoptosis, the flow cytometric analysis was performed. The proportion of heat stress-induced apoptosis decreased from 56 to 17% by heat shock pretreatment that induces no apoptosis (Figs. 1B, 1C). Moreover, the activity of caspases was measured using a fluorescence substrate.16) The caspase activity elevated by heat stress was greatly inhibited by heat shock, as seen in DNA fragmentation (Fig. 1D). These results suggest that the apoptosis induced by heat stress in MEL cells is suppressed by pretreatment of heat shock.
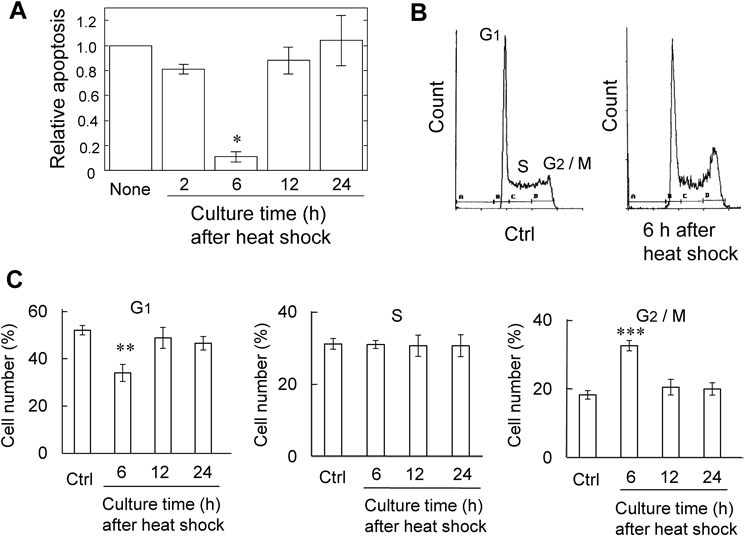
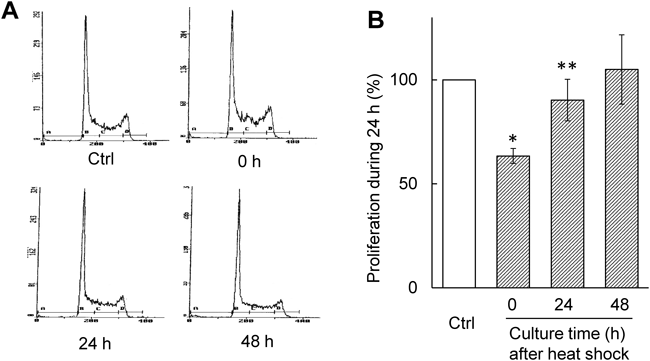
Culture for 6 h after Heat Shock Induces Thermotolerance in MEL CellsWe examined the effect of culture time between the heat shock and heat stress on heat stress-induced apoptosis. The MEL cells subjected to the heat shock were cultured for 2, 6, 12, and 24 h prior to heat stress. The heat stress-induced apoptosis was greatly suppressed upon 6 h culture after heat shock (Fig. 2A). However, such suppressive effect was disappeared at 12 h after heat shock (Fig. 2A). To explain this result, cell cycles of MEL cells subjected to heat shock were examined using a flow cytometry (Fig. 2B). Upon 6 h culture after heat shock, the number of cells in G1 phase decreased, whereas G2/M cell populations increased (Figs. 2B, 2C). After 12 and 24 h, however, the cell population in each phase was restored to the original level (Fig. 2C). Moreover, the proliferation rate of MEL cells was examined using cells cultured for 0, 24, and 48 h after heat shock. The proliferation ability was estimated as the cell populations that increased during 24 h culture. At 24 h after heat shock, the proliferation ability as well as the cell cycle restored to the original level (Fig. 3). Thus, upon 6 h culture after heat shock MEL cells arrest in G2 phase and acquire thermotolerance, but these properties are abrogated upon culture over 12 h.
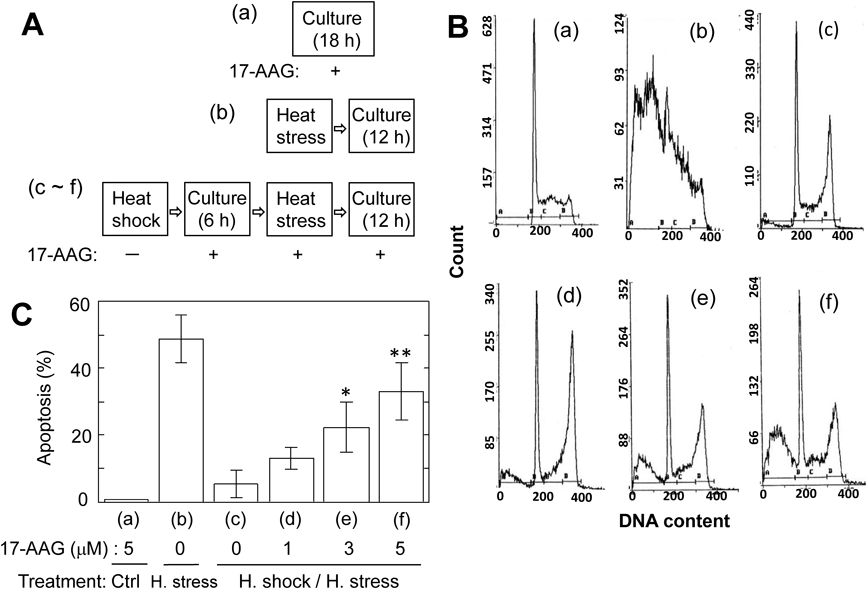
Suppressive Effects of Heat Stress-Induced Apoptosis by Heat Shock Are Abrogated in the Presence of HSP90 Inhibitor, 17-AAGIn most cells, HSP72 is known to be induced upon exposure to heat shock.10,18) In MEL cells, however, no expression of HSP72 by heat shock for 10 min at 44°C was observed (Fig. S1), as mentioned in previous works.11,19) A number of studies report that HSP90 also plays an important role in mammalian cells exposed to physicochemical stresses such as heat and UV.11,20) So, we examined using 17-AAG,12,21) a specific inhibitor of HSP90, the contribution of HSP90 concerning behaviors of MEL cells exposed to heat shock or/and heat stress (Figs. 4A–4C). When intact MEL cells were cultured for 18 h in the presence of 5 µM 17-AAG, no apoptosis was observed (a). As mentioned above, apoptosis was observed upon 12 h culture of MEL cells exposed to heat stress (b). When MEL cells exposed to heat shock were subjected in the presence of 17-AAG to 6 h culture, heat stress, and then 12 h culture, the proportion of apoptosis increased upon depending on the concentration of 17-AAG (c–f). This indicates that the suppressive effects of apoptosis by heat shock are abrogated by 17-AAG. Novobiocin,12,22) another inhibitor of HSP90, also abolished such effects by heat shock (Fig. S2). Thus, the results from HSP90 inhibitors strongly suggest that HSP90 might be involved in the suppression of heat stress-induced apoptosis by heat shock.
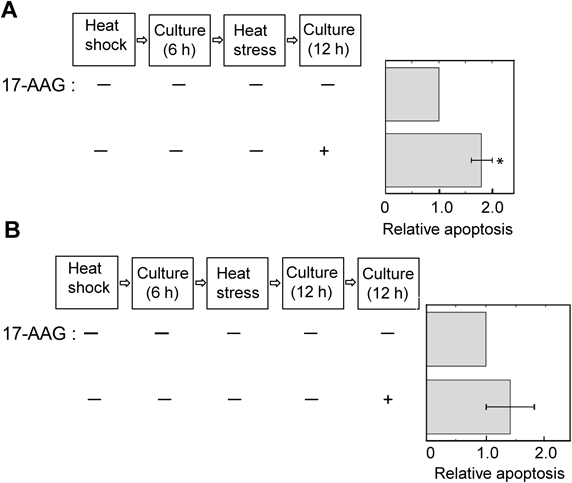
Time Course of Suppression by Heat Shock of Heat Stress-Induced ApoptosisTo examine how long the suppressive effects by heat shock persist, the addition time of 17-AAG to the MEL cell suspensions was changed. When heat stress-exposed MEL cells, which were pretreated upon 6 h culture after heat shock, were cultured for 12 h in the presence of 17-AAG, the proportion of apoptosis increased significantly, compared with that in the absence of 17-AAG (Fig. 5A). On the other hand, such heat stress-exposed MEL cells were cultured for 12 h in the absence of 17-AAG, and the cells diluted with fresh medium due to their proliferation were cultured furthermore for 12 h in the presence of 17-AAG. In this case, no enhancement of apoptosis was observed by 17-AAG (Fig. 5B). This suggests that the suppressive effects of heat stress-induced apoptosis by heat shock are abolished upon culture beyond 18 h after heat shock.
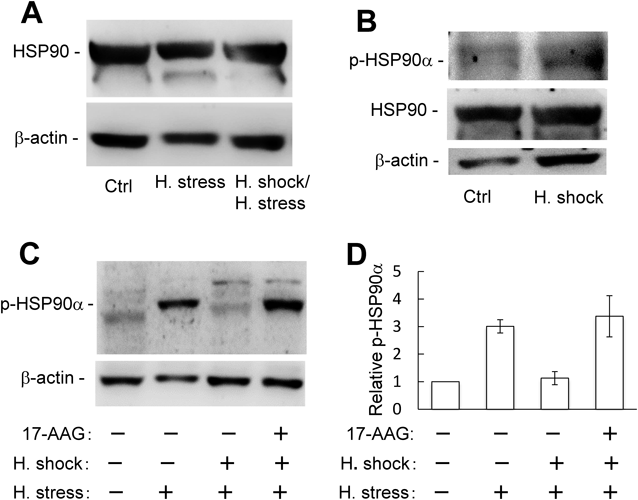
Phosphorylation of HSP90α Induced by Heat Stress Is Inhibited by Heat Shock PretreatmentWhen the MEL cells exposed to heat stress were cultured for 12 h, the high expression level of HSP90 was almost the same as that of the stress-free cells (Fig. 6A). Similarly, such expression level of HSP90 remained constant in the MEL cells that heat stress-induced apoptosis was suppressed by the pretreatment of heat shock (Fig. 6A). At 6 h after heat shock, moreover, the expression levels of HSP90 did not change and the phosphorylation of HSP90α (p-HSP90α) was not detected (Fig. 6B). On the other hand, the band of p-HSP90α was observed in the MEL cells exposed to heat stress, but not in cells pretreated with heat shock prior to heat stress (Fig. 6C). Interestingly, the level of p-HSP90α increased when heat stress-exposed MEL cells, which are pretreated upon 6 h culture after heat shock, were cultured for 12 h in the presence of 17-AAG (Figs. 6C, 6D). These results suggest that the phosphorylation of HSP90α is associated with apoptosis in MEL cells.
As mentioned above, the expression level of HSP90 in MEL cells was unaffected by heat shock. So, the expression of survivin23) and STK38,24,25) which are client proteins of HSP90, was examined by Western blotting. However, their expression levels remained unaltered at 6 h after heat shock (Fig. S3).
JNK Pathway Is Activated in Heat Stress-Induced ApoptosisIt is reported that JNK is activated in MEF cells exposed to heat stress and results in apoptosis.10) So, we examined whether the JNK pathway is activated by heat stress in MEL cells. In MEL cells exposed to heat stress, the band of p-JNK was observed (Fig. 7). However, no such a band appeared in the cells subjected to heat shock before heat stress (Fig. 7). These results suggest that the JNK pathway in MEL cells is activated in the heat stress-induced apoptosis.
DISCUSSION
In the present work, we have demonstrated that MEL cells are capable of acquiring thermotolerance if they are subjected to heat shock followed by culturing for several hours. Thus, heat stress-induced apoptosis is suppressed by heat shock pretreatment. Such suppressive effects are maximal when the MEL cells are cultured for 6 h after heat shock. Similar results are reported in MEF and human lymphoid cells such as U937 and PEER.10,18) In these cells, the expression level of HSP72 increases at 6 h after heat shock and heat stress-induced apoptosis is significantly suppressed.10,18) Here, HSP72 plays an important role in the suppression of heat stress-induced apoptosis. On the other hand, it is of importance to note that HSP72 is deficient in MEL cells.11) However, HSP90 is highly expressed in MEL cells. In the case of MEL cells, the suppression of heat stress-induced apoptosis by heat shock is abrogated by specific inhibitors of HSP90. Interestingly, the phosphorylation of HSP90α is observed in heat stress-induced apoptotic MEL cells. Such phosphorylation is inhibited in MEL cells that the apoptosis is suppressed by heat shock, but restored in the presence of 17-AAG (Figs. 6C, 6D). Therefore, it seems likely that the phosphorylation of HSP90α is involved in the apoptosis.26,27) These results suggest that HSP90α responds to extracellular stress such as heating in MEL cells.
Generally, the progression of cell cycle reflects the response of cells to external perturbation. In this work, upon 6 h culture after heat shock, cells in G2/M phase increase, whereas cells in G1 phase decrease. This indicates that the progression of cell cycle is arrested at G2 phase. However, normal cell cycle is observed upon 12 h culture after heat shock. Moreover, the proliferation rate of cells also supports these results (Fig. 3). Thus, the cell cycle and proliferation rate of MEL cells are restored to the original state at 12 h after heat shock. These results explain the effects of culture time after heat shock on apoptosis, as seen in Fig. 2A.
Heat stress-induced apoptosis in MEL cells is suppressed by heat shock pretreatment. Such suppressive effects are abrogated by HSP90 inhibitors such as 17-AAG and novobiocin so that heat stress-induced apoptosis is enhanced. Thus, we can check such suppressive effects by heat shock against heat stress-induced apoptosis using HSP90 inhibitors, as shown in Fig. 5. HSP90 is highly overexpressed in cancer cells.12) In the present work, it has found that HSP90 is abundantly expressed in MEL cells. Therefore, it seems unnecessary for MEL cells to increase the expression level of HSP90 after heat shock. HSP90 consists of three domains.12) 17-AAG interacts with the amino terminal domain which is an ATP-binding pocket so that it prevents the chaperone activity of HSP90.12) On the other hand, novobiocin inhibits HSP90 activity by binding to its carboxy-terminal domain.12) Thus, the results from 17-AAG and novobiocin in the present work suggest that HSP90 activity contributes to the suppressive effects by heat shock of apoptosis in MEL cells.
HSP90 is phosphorylated by kinases such as casein kinase II28) and Wee1.29) Chaperon functions of HSP90 are affected by its phosphorylation. In response to DNA-damaging agents, HSP90α is phosphorylated by DNA-dependent protein kinase (DNA-PK).26) Phosphorylated HSP90α is accumulated in repair foci.27) On the other hand, the apoptotic ring that is characterized by the phosphorylation of histone H2AX is observed in cells treated with apoptotic agents such as camptothecin and staurosporine.26) This apoptotic ring contains DNA damage response proteins such as activated check point kinase 2 (Chk2) and activated ataxia telangiectasia mutated (ATM). Interestingly, p-HSP90α and phosphorylated DNA-PK also are involved in such apoptotic ring.26) HSP90α is phosphorylated by activated DNA-PK on threonines 5 and 7.26) Using antibody that can recognize the phosphorylated threonines 5 and 7 on HSP90α, we were able to detect the band of p-HSP90α in MEL cells that apoptosis is induced by heat stress. However, such phosphorylation of HSP90α is inhibited by heat shock pretreatment, but restored by 17-AAG addition after heat shock. Thus, HSP90α is phosphorylated in heat stress-induced apoptotic MEL cells, perhaps by activated DNA-PK.
First, when MEL cells were cultured for 6 h at 37°C after heat shock, we expected the increment of expression level of HSP90 because the thermotolerance effect by heat shock is abrogated by 17-AAG. However, the HSP90 expression level remained constant during the culture after heat shock, in contrast to the case of HSP72 in MEF.10) So, we focused the client proteins of HSP90. Survivin is a client protein of HSP90 and most popular anti-apoptotic player.23) However, the low level of survivin was unaffected by heat shock. Recently, it has been reported that 17-AAG-treated HeLa cells reveal a reduced level of cellular STK38 and become more sensitive to oxidative stress such as X-ray irradiation.24) Here, cellular STK38 level is modulated by SP1, a transcription factor and client protein of HSP90.24) In this work, cellular STK levels in MEL cells were unaffected by culture time after heat shock, indicating no contribution of STK38 to the suppression of heat stress-induced apoptosis. Of client proteins of HSP90, Akt is an important player in the cell survival signaling pathway.8,30) Activated Akt phosphorylates Bad so that the functions of Bad as a facilitator of apoptosis are lost.31) Therefore, the inhibition of HSP90 by 17-AAG results in the reduction of intracellular level of Akt.31) Thus, apoptosis is facilitated by inhibiting the survival signal. We expect that the investigation about the behavior of Akt after heat shock provides useful information on acquiring thermotolerance described here.
MAPKs are activated for some physicochemical stresses.14,32) Three major groups of MAPKs in mammals have been identified as p38, JNKs, and extracellular signal regulated kinases (ERKs)32) Activation of the ERK signal pathway leads to cell survival, whereas cell death such as apoptosis is induced by the activated JNK or p38 pathway.7,14) Thus, the fate of cells is determined depending on which pathway is activated by the degrees and properties of cell damages. In mammalian cells exposed to heat stress, the ERK signal transduction pathway is activated.2) However, leukemia cells such as HL-60 and U-937 cells lack the activation of Raf-1 so that the ERK pathway fails to be activated by heat stress.2,18) Thus, these leukemia cells containing MEL cells are more sensitive to heat stress.2) In the present work, the phosphorylation of JNKs has been observed in heat stress-induced apoptotic MEL cells. However, such an activation of JNKs is inhibited in MEL cells that heat stress-induced apoptosis is suppressed by heat shock pretreatment. Thus, the JNK pathway is activated in heat stress-induced apoptotic MEL cells. Further study is necessary concerning the contribution of HSP90 to JNK pathway in MEL cells.
In conclusion, we have demonstrated that MEL cells exhibit the most effective effect on the thermotolerance at 6 h after heat shock, but lose such property after 12 h. Results from HSP90 inhibitors such as 17-AAG and novobiocin reveal that HSP90 activities are associated with such thermotolerance in HSP72-deficient MEL cells. Moreover, Western blotting shows that HSP90 in MEL cells is expressed at high level and HSP90α is phosphorylated under apoptosis. Thus, HSP90α plays an important role via its client protein in the acquirement of thermotolerance. Moreover, the present work demonstrates that geldanamycin analogues containing 17-AAG are useful drugs capable of inducing apoptosis in the cells whose HSP72 expression is deficient and HSP90α is activated by extracellular stress such as anticancer drugs and heating.
Acknowledgment
This work was supported in part by a Grant (155003) from the Central Research Institute of Fukuoka University.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Kemp MG, Spandau DF, Simman R, Travers JB. Insulin-like growth factor 1 receptor signaling is required for optimal ATR-CHK1 kinase signaling in ultraviolet B (UVB)-irradiated human keratinocytes. J. Biol. Chem., 292, 1231–1239 (2017).
- 2) Woessmann W, Meng Y-H, Mivechi NF. An essential role for mitogen-activated protein kinases, ERKs, in preventing heat-induced cell death. J. Cell. Biochem., 74, 648–662 (1999).
- 3) Yamaguchi T, Hashiguchi K, Katsuki S, Iwamoto W, Tsuruhara S, Terada S. Activation of the intrinsic and extrinsic pathways in high pressure-induced apoptosis of murine erythroleukemia cells. Cell. Mol. Biol. Lett., 13, 49–57 (2008).
- 4) Zunino B, Rubio-Patiño C, Villa E, Meynet O, Proics E, Cornille A, Pommier S, Mondragón L, Chiche J, Bereder JM, Carles M, Ricci JE. Hyperthermic intraperitoneal chemotherapy leads to an anticancer immune response via exposure of cell surface heat shock protein 90. Oncogene, 35, 261–268 (2016).
- 5) Samali A, Cotter TG. Heat shock proteins increase resistance to apoptosis. Exp. Cell Res., 223, 163–170 (1996).
- 6) Landry J, Lambert H, Zhou M, Lavoie JN, Hickey E, Weber LA, Anderson CW. Human HSP27 is phosphorylated at serines 78 and 82 by heat shock and mitogen-activated kinases that recognize the same amino acid motif as S6 kinase II. J. Biol. Chem., 267, 794–803 (1992).
- 7) Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell. Biol., 15, 505–516 (1995).
- 8) Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol., 18, 345–360 (2017).
- 9) Khalil AA, Kabapy NF, Deraz SF, Smith C. Heat shock proteins in oncology: diagnostic biomarkers or therapeutic targets? Biochim. Biophys. Acta, 1816, 89–104 (2011).
- 10) Buzzard KA, Giaccia AJ, Killender M, Anderson RL. Heat shock protein 72 modulates pathways of stress-induced apoptosis. J. Biol. Chem., 273, 17147–17153 (1998).
- 11) Hensold JO, Hunt CR, Calderwood SK, Housman DE, Kingston RE. DNA binding of heat shock factor to the heat shock element is insufficient for transcriptional activation in murine erythroleukemia cells. Mol. Cell. Biol., 10, 1600–1608 (1990).
- 12) Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer, 10, 537–549 (2010).
- 13) Kondo T, Matsuda T, Kitano T, Takahashi A, Tashima M, Ishikura H, Umehara H, Domae N, Uchiyama T, Okazaki T. Role of c-jun expression increased by heat shock- and ceramide-activated caspase-3 in HL-60 cell apoptosis. Possible involvement of ceramide in heat shock-induced apoptosis. J. Biol. Chem., 275, 7668–7676 (2000).
- 14) Yabu T, Shiba H, Shibasaki Y, Nakanishi T, Imamura S, Touhata K, Yamashita M. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ., 22, 258–273 (2015).
- 15) Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer, 13, 51–65 (2013).
- 16) Take J, Yamaguchi T, Mine N, Terada S. Caspase activation in high-pressure-induced apoptosis of murine erythroleukemia cells. Jpn. J. Physiol., 51, 193–199 (2001).
- 17) Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685 (1970).
- 18) Gabai VL, Meriin AB, Mosser DD, Caron AW, Rits S, Shifrin VI, Sherman MY. Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J. Biol. Chem., 272, 18033–18037 (1997).
- 19) Hensold JO, Housman DE. Decreased expression of the stress protein HSP70 is an early event in murine erythroleukemic cell differentiation. Mol. Cell. Biol., 8, 2219–2223 (1988).
- 20) Sekimoto T, Oda T, Pozo FM, Murakumo Y, Masutani C, Hanaoka F, Yamashita T. The molecular chaperone Hsp90 regulates accumulation of DNA polymerase η at replication stalling sites in UV-irradiated cells. Mol. Cell, 37, 79–89 (2010).
- 21) Porter JR, Fritz CC, Depew KM. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr. Opin. Chem. Biol., 14, 412–420 (2010).
- 22) Jego G, Hazoumé A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett., 332, 275–285 (2013).
- 23) Fortugno P, Beltrami E, Plescia J, Fontana J, Pradhan D, Marchisio PC, Sessa WC, Altieri DC. Regulation of survivin function by Hsp90. Proc. Natl. Acad. Sci. U.S.A., 100, 13791–13796 (2003).
- 24) Enomoto A, Fukasawa T, Takamatsu N, Ito M, Morita A, Hosoi Y, Miyagawa K. The HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin modulates radiosensitivity by downregulating serine/threonine kinase 38 via Sp1 inhibition. Eur. J. Cancer, 49, 3547–3558 (2013).
- 25) Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, Hemmings BA. NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr. Biol., 18, 1889–1895 (2008).
- 26) Solier S, Kohn KW, Scroggins B, Xu W, Trepel J, Neckers L, Pommier Y. Heat shock protein 90α (HSP90α), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc. Natl. Acad. Sci. U.S.A., 109, 12866–12872 (2012).
- 27) Quanz M, Herbette A, Sayarath M, de Koning L, Dubois T, Sun JS, Dutreix M. Heat shock protein 90α (Hsp90α) is phosphorylated in response to DNA damage and accumulates in repair foci. J. Biol. Chem., 287, 8803–8815 (2012).
- 28) Lees-Miller SP, Anderson CW. Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J. Biol. Chem., 264, 2431–2437 (1989).
- 29) Mollapour M, Tsutsumi S, Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle, 9, 2310–2316 (2010).
- 30) Georgakis GV, Li Y, Rassidakis GZ, Martinez-Valdez H, Medeiros LJ, Younes A. Inhibition of heat shock protein 90 function by 17-allylamino-17-demethoxy-geldanamycin in Hodgkin’s lymphoma cells down-regulates Akt kinase, dephosphorylates extracellular signal-regulated kinase, and induces cell cycle arrest and cell death. Clin. Cancer Res., 12, 584–590 (2006).
- 31) Gao Z, Liu F, Yin P, Wan C, He S, Liu X, Zhao H, Liu T, Xu J, Guo S. Inhibition of heat-induced apoptosis in rat small intestine and IEC-6 cells through the AKT signaling pathway. BMC Vet. Res., 9, 241 (2013).
- 32) Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell, 103, 239–252 (2000).