Abstract
Peroxisomes are indispensable organelles in mammals including humans. They are involved in the β-oxidation of very long chain fatty acids, and the synthesis of ether phospholipids and bile acids. Pre-peroxisomes bud from endoplasmic reticulum and peroxisomal membrane and matrix proteins are imported to the pre-peroxisomes. Then, matured peroxisomes grow by division. Impairment of the biogenesis and function of peroxisomes results in severe diseases. Since I first undertook peroxisome research in Prof. de Duve’s laboratory at Rockefeller University in 1985, I have continuously studied peroxisomes for more than 30 years, with a particular focus on the ATP-binding cassette (ABC) transporters. Here, I review the history of peroxisome research, the biogenesis and function of peroxisomes, and peroxisome disease including X-linked adrenoleukodystrophy. The review includes the targeting and function of the ABC transporter subfamily D.
1. INTRODUCTION
Peroxisomes are single membrane-bound organelles existing in almost all eukaryotes and ubiquitous in mammalian cells. Most cells have spherical or spheroidal peroxisomes with a diameter 0.1–1 µm although the peroxisomes vary in shape and size in different tissues (Fig. 1). Peroxisomes are small organelles that comprise only a few percent of the volume of the human liver and kidney cells, even though peroxisomes are known to be abundant in these cells. Nonetheless, despite the low abundance, peroxisomes perform essential functions.
These organelles were first described in 1954 by Rhodin, a postgraduate student at the Karolinska Institute working in the laboratory of Sjöstrand, one of the pioneers of electron microscopy. Rhodin was carefully examining the proximal tubule cells of the mouse kidney and observed a small organelle that had not been previously described. It was surrounded by a single membrane and filled with a fine granular matrix. He must have been excited about the appearance of an unknown cell compartment he called “microbodies.”1,2) However, researchers had not as yet expressed any interest in these microbodies since their function was unknown.
The microbodies were characterized biochemically by de Duve and colleagues in 1965.3) de Duve had hypothesized that all members of a given organelle have the same enzymatic composition. He undertook efforts to separate the microbodies from other organelles, especially the lysosomes. Initially, the distribution of the microbodies (indicated by the presence of urate oxidase, a marker enzyme) and lysosomes (acid phosphatase) were located together when normal rat liver cells were fractionated. In contrast, when rats were injected with the detergent Triton WR-1339, the lysosome density was selectively lowered by the accumulation of lipids derived from plasma lipoproteins, and the particles containing urate oxidase were clearly separated from the lysosomes.4) They also found that microbodies contained various H2O2-producing oxidases and H2O2-degrading enzyme catalase.5) They named the organelles “peroxisomes,” but their physiological function was still poorly understood.
In the 1970s, peroxisomes were suggested to be involved in lipid metabolism due to evidence that clofibrate and other hypolipidemic drugs induced proliferation of hepatic peroxisomes.6) In 1976, Lazarow and de Duve found that rat liver peroxisomes were capable of palmitoyl-CoA oxidation with reduction of O2 to H2O2 in a cyan-insensitive manner and that this oxidation was enhanced in rats given the peroxisome proliferator clofibrate.7) This was the first report on the physiological function of peroxisomes in mammals. They also suggested that the hepatic fatty acid β-oxidation system plays a role in reducing serum levels of neutral lipids by hypolipidemic drugs. It was subsequently shown that treating rodents with these drugs also induced certain peroxisomal enzymes involved in fatty acid β-oxidation.8)
In terms of peroxisomal disease, Bowen first indicated that Zellweger’s cerebro-hepato-renal syndrome could be considered a prototype of genetic disorders in 1964.9) Goldfisher subsequently discovered that peroxisomes were absent from tissues in Zellweger’s patients.10) It was soon recognized that peroxisomes are essential for human health and development.
Peroxisomes are essential for lipid metabolism and play a critical role in the homeostasis of multiple organs. Peroxisomes in all neural cell types are smaller than those in liver and kidney cells, but are nonetheless important for proper neurological development.11) The indispensable role of peroxisomes in human health and development is evidenced by the existence of a large number of inborn errors of peroxisome assembly and peroxisomal metabolism leading to severe diseases.12) At present, 23 genetic diseases caused by mutations of 32 known genes are classified as peroxisome disease.
Peroxisomes are highly dynamic organelles and can rapidly change in size, abundance, and protein content in response to alterations of nutritional and environmental conditions. In rodents and humans, physiological stimuli such as diabetes mellitus and high-fat diets induce peroxisomal proliferation.13)
Glycosomes, glyoxysomes and Woronin bodies are known as organelles related to peroxisomes. They are formed by the same mechanism as peroxisomes and carry out specific functions in different species. Glycosomes in infectious protozoa possess enzymes for glycolysis in the host animal blood stream, including humans.14) Glyoxysomes include fatty acid β-oxidation and glyoxylate cycle enzymes, and are involved in germination in plants.15) Woronin bodies in ascomycete fungi contain a structural protein, hexagonal peroxisome protein (Hex1). The hyphal cells are separated by perforated septa by Woronin bodies. They seal the pore to protect the remaining cells when extensive wound-induced damage occurs.16)
2. BIOGENESIS AND FUNCTION OF PEROXISOMES
2.1. Historical StudiesIn the 1960–70s, peroxisomes were thought to be formed from the endoplasmic reticulum (ER), since they often appeared in clusters surrounded by a smooth ER and seemed to be continuous with the ER in a number of places.17) A pulse-chase study of catalase in the rat liver had shown that labeled-catalase appeared first in the ER fraction then was transported to the mitochondrial fraction,18) although peroxisomes had not been identified in tissue. In the 1980s, it was demonstrated that many peroxisomal proteins, including membrane proteins, are synthesized on free polysomes. In vitro import studies were developed that showed that newly synthesized peroxisomal matrix proteins were directly transported into peroxisomes. We demonstrated that acyl-CoA oxidase, a peroxisomal matrix protein, was imported into purified rat liver peroxisomes and that the import required ATP hydrolysis, but not a change in the membrane potential.19) At the time, it had been generally accepted that peroxisomes could form by growth and division from pre-existing organelles, as do mitochondria.20)
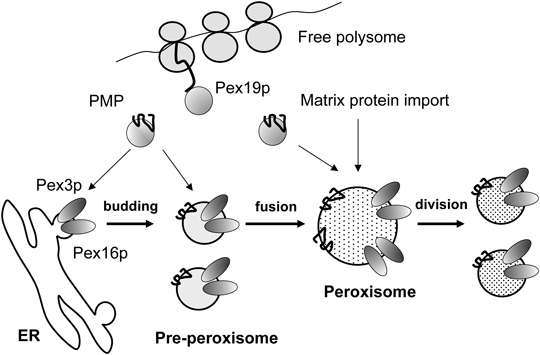
Since the 1990s, many of the genes involved in the biogenesis of peroxisomes have been identified. (The gene is called “PEX” which encodes peroxin, Pexp for short.) It was shown that peroxisomes were not detected in cells with a mutation of the PEX3 or PEX19 gene, and peroxisomes appeared upon transfection with a normal cDNA of PEX3 or PEX19, respectively. Furthermore, newly synthesized Pex3p was shown to be inserted into ER membranes. Based on these observations, it is currently considered most likely that peroxisomes are ER-derived organelles in mammals,21) although there is a suggestion that pre-peroxisomes are formed by the fusion of vesicles derived from the ER and mitochondrial outer membranes.22) Biosynthesis of peroxisomes in mammals involves three different processes: budding of pre-peroxisomes from ER, the import of peroxisomal membrane proteins (PMPs) and matrix proteins to the pre-peroxisomes and peroxisomes, growth to mature peroxisomes, and fission (Fig. 2).
In retrospect, the model of peroxisome biogenesis was proposed based on the experimental results obtained by the latest technology at that time, and then the model was reevaluated and revised based on new evidence obtained by subsequently emerging technology. Tracing the evolving history of the concept of the biogenesis of peroxisome is interesting. The idea of where the peroxisome formed goes back to the ER hypothesis from over half a century ago. It is known that many of the so-called peroxins encoded by PEX genes are involved in peroxisome biogenesis. To date, 36 peroxins have been identified. Among them, the mutations of 13 peroxins are identified as the causative of peroxisome biogenesis disorder (PBD) with a severe phenotype.
With regard to peroxisome function, several different metabolic pathways, including very long chain fatty acid (VLCFA) β-oxidation, as well as the synthesis of ether-phospholipids and bile acids, were characterized after the β-oxidation system had been reported in mammalian peroxisomes.7) Elucidation of these metabolic roles of peroxisomes in humans is coupled to research on Zellweger syndrome, in which no functional peroxisome exists. In the middle-1980s, Moser et al. demonstrated that VLCFA was increased in plasma and cultured skin fibroblasts from the patients, and oxidation of VLCFA was impaired in the homogenates of cultured skin fibroblasts.23) Activity of dihydroxyacetone phosphate acyltransferase (DHAPAT), a peroxisomal enzyme with a major role in ether lipid synthesis, was found to be deficient in the skin fibroblasts of patients with Zellweger syndrome.24) Defective peroxisomal cleavage of the C27-steroid side chain in bile acid synthesis was also found in Zellweger syndrome.25) Later, antibodies against peroxisomal enzymes prepared by Hashimoto’s group at Shinshu University helped to confirm several peroxisomal diseases associated with deficiency in the peroxisomal β-oxidation enzymes.
2.2. Targeting of Peroxisomal Matrix Proteins to PeroxisomesBecause peroxisomes do not contain any DNA, all of the peroxisomal proteins are encoded by genomic DNA. Several in vitro import studies including our own have suggested that peroxisomal matrix proteins are posttranslationally imported into peroxisomes.19) In addition, in 1987 Gould, then a postgraduate student at the laboratory of Subramini at University of California (San Diego), found a peroxisome targeting signal in the COOH-terminal 12 amino acids of firefly luciferase by the transfection of the cDNA in CV-1 monkey cells (the luciferase is present in peroxisome-like organelles in the cells of the firefly lantern organ).26) He subsequently showed that the COOH-terminal amino acids Ser-Lys-Leu (SKL) were sufficient for the targeting of the protein to peroxisomes.27) In contrast, peroxisomal 3-ketoacyl CoA thiolase was found to possess a peroxisomal targeting signal at the NH2-terminus as a pre-sequence. This sequence is removed after the import of peroxisomes.28,29)
Subsequently, cytosolic peroxisomal targeting signal receptors Pex5p and Pex7p were identified. It was shown that a complex of a peroxisomal protein with its receptor is transported into peroxisomes through pores on the peroxisomal membrane, and then the receptor is recycled in the cytosol (Fig. 3A).
The correct targeting of peroxisomal matrix proteins is mediated by a specific targeting sequence known as the peroxisome targeting signal (PTS). There are two kinds of targeting sequences, so-called PTS1 and 2. PTS1 is the tripeptide (S/A/C)-(K/R/H)-(L/M) at COOH-terminus of the majority of the peroxisomal matrix proteins. On the other hand, PTS2 is a nonapeptide consisting of the consensus sequence (R/K)-(L/I/K)-X5-(Q/H)-(L/I/V) at the NH2-terminus of a few proteins. Newly synthesized matrix proteins containing PTS1 are recognized by the cytosolic protein Pex5p. There are two types of Pex5p in mammalian cells, Pex5pL and Pex5pS. They are produced by the differential splicing of the PEX5 gene. Pex7p recognizes the NH2-terminal PTS2. Pex7p needs to interact with Pex5pL to be targeted to peroxisome (Fig. 3A). The matrix protein-loaded receptor proteins dock onto the peroxisomal docking complex constituted by an oligomer of Pex14p and Pex13p. The docking complex also interacts with another complex of Pex2p, Pex10p and Pex12p. After docking, the matrix protein-loaded receptor is translocated to the inside of the peroxisomal membrane. Then, the matrix proteins are released into the peroxisomal lumen and the receptor proteins (Pex5p and Pex7p) return to the cytosol for another import cycle or are directed to the proteasome for degradation.30,31) These latter two steps require ubiquitin ligases activities of Pex2p, Pex10p and Pex12p that catalyze either the mono- or poly-ubiquitination of Pex5p. Mono-ubiquitination promotes the recycling of Pex5p, whereas poly-ubiquitination renders Pex5p a substrate for proteasome-mediated degradation.32) Pex1p and Pex6p are members of ATPases associated with diverse cellular activities (AAA ATPase). They form a complex as a trimer of Pex1p/Pex6p heterodimer and interact with Pex26p on peroxisomal membranes. The Pex1p-Pex6p-Pex26p complex is involved in the release of ubiquitinated Pex5p from the peroxisomal membrane.33,34)
The mechanism of peroxisomal matrix protein transport into peroxisomes is quite unique. The cytosolic receptors are required for targeting and the receptor complex with the peroxisomal protein translocates through the peroxisomal pore that transitionally opens and closes. This sort of sophisticated system is required for the correct and efficient transport of newly synthesized peroxisomal proteins to peroxisomes because of the very narrow area afforded by the peroxisomal membranes in the cells.
2.3. Biogenesis of the Peroxisomal MembraneIn the 1980s, it was thought that the peroxisomes did not exist in the cells of patients with Zellweger syndrome. We prepared antibodies against PMPs and endeavored to characterize PMP behavior in Zellweger syndrome fibroblasts. Interestingly, several PMPs were detected in the fibroblasts and they sedimented at a density of 1.18 on sucrose gradient centrifugation. In addition, they were separated from other organelle proteins, suggesting that PMPs did not mistarget to other organelles, but rather, localized to some sort of particles. We showed that peroxisomal membrane particles are present in fibroblasts of Zellweger syndrome through immunofluoresent as well as immunoelectron microscopy and named the particles “peroxisome ghosts” since they did not include matrix proteins.35,36) These results suggested that the mechanism by which PMPs target peroxisomes is different from that of peroxisome matrix proteins.37)
Then several studies including our own showed that the targeting signal of peroxisomal membrane proteins (mPTS, there is no constant amino acid sequence) is different from that of PTS1 and PTS2. A different set of peroxins has been reported to be required for this process in other mammals. Pex19p, Pex3p, and Pex16p are involved in the membrane assembly and insertion of PMPs (Fig. 3B).
Pex19p is mainly a cytosolic protein and is capable of binding many different PMPs. It has been postulated to serve as the receptor for newly synthesized PMPs.38) PMPs are bound to Pex19p via the mPTS in their sequences. Pex19p is a chaperone-like protein that keeps PMP soluble in cytosol and inserts PMP into the peroxisomal membrane through the interaction with Pex3p. Pex3p is located in the peroxisomal membrane and serves as a docking site for PMP-loaded Pex19p.
To understand the molecular mechanism by which PMP is inserted into the peroxisomal membrane, Kato and colleagues at Kyoto University attempted to crystalize Pex19p. Purified human Pex19p expressed in Escherichia coli was highly soluble, and did not crystalize at a concentration over 50 mg/mL. We found Pex19p binds to Pex3p at the NH2-terminal 44 amino acids and to PMP at the COOH-terminal domain after the 91st amino acid.39) Then Kato’s group succeeded in crystalizing the NH2-terminal 44 amino acids of Pex19p complexed with the cytosolic domain of Pex3p (aa.49-373).40) The overall shape of the Pex3p structure was a prolate spheroid with a novel antiparallel helical fold. The Pex19p-binding site on Pex3p was at one apex of the spheroid near Trp104. A 16-residue region of the Pex19p peptide formed an α-helix and made contact with Pex3p; this helix was disordered in the unbound state. The Pex19p peptide contained a characteristic motif, consisting of the leucine triad (Leu18, Leu21, Leu22), and Phe29, which together were critical for Pex3p binding (Fig. 3B). When the wild-type Pex19p or the L26A mutant Pex19p fused with green fluorescent protein (GFP) was expressed in PEX19-deficient cells, punctate peroxisomes were detected in the GFP-stained cells using anti-PMP70/ABCD antibodies. On the other hand, the L22A and F29A mutants did not restore any peroxisomal structures in the cells. Therefore these interactions are evidently important for the assembly of the peroxisome in vivo. The dissociation constant of Pex19p with Pex3p in vitro was almost the same at the nM level with or without PMP on Pex19p,41) suggesting that another protein must be required for the dissociation of Pex19p to Pex3p after Pex19p unloads PMP on the peroxisomal membrane. In addition, the COOH-terminal domain was shown to be involved in the binding of PMPs, and farnesylation at the COOH-terminal CaaX motif enhances the PMP interaction.42)
Pex16p was shown to serve as the receptor for Pex19p loaded with newly synthesized Pex3p.43) Pex16p functions as a tethering factor for Pex3p and seems to be a part of the putative membrane-insertion machinery. It has been reported that Pex3p and Pex16p localize to the ER, suggesting that pre-peroxisomes may be derived from the ER (Fig. 2). Dysfunction of Pex3p, Pex16p or Pex19p results in the absence of peroxisomes, which in turn causes in Zellweger syndrome.
2.4. Functions of PeroxisomesPeroxisomes play essential roles in cellular functions and are involved in various metabolic pathways unique to this organelle.12) In terms of peroxisome function, we found lon protease that is involved in the quality control of peroxisomal proteins by proteome analysis of rat liver peroxisomes.44) In addition, we reported that insulin degrading enzyme with PTS1 is located in part in peroxisomes and involved in the degradation of oxidized proteins instead of insulin.45) We also found an ATP/AMP carrier on peroxisomal membrane in yeast cells.46)
Among the metabolic processes in peroxisomes, the β-oxidation of fatty acids is the major catabolic pathway. The following fatty acids are oxidized only in peroxisomes: saturated and polyunsaturated VLCFA, long-chain dicarboxylic acids, 3 or 2-metyl branched-chain fatty acids such as phytanic acid (3,7,11,15-tetrametylhexadeconoic acid) and pristanic acid (2,6,10,14-tetrametylpentadeconoic acid) as well as bile acid intermediates (dihydroxycholestanoic acid; DHCA and trihydroxycholestanoic acid; THCA) (Fig. 4). These fatty acids are imported into peroxisomes in the CoA form via the ATP-binding cassette (ABC) transporters ABCD1–3, degraded by one or more cycles of β-oxidation, released into the cytosol, and transported to mitochondria for further metabolism.47) 25R-DHCA/THCA-CoA is converted to 25S-DHCA/THCA-CoA by 2-metyl-acyl-CoA racemase. Phytanoyl-CoA exist as 3R and 3S forms and these are first converted to 2R- and 2S-pristanoyl-CoA by α-oxidation since β-methyl branched chain of phytanic acid cannot undergo β-oxidation. Then 2R-pristanoyl-CoA is converted to 2S-pristanoyl-CoA by 2-metyl-acyl-CoA racemase.
As is the case in mitochondria, the β-oxidation in peroxisomes is conducted via four steps (Fig. 4). In peroxisomes, electrons from FAD in acyl-CoA oxidase (ACOX) are transferred directly to O2 to generate H2O2, which is degraded by catalase into H2O and O2, while re-oxidation of FADH2 is coupled to the electron transport chain so as to produce ATP in mitochondria.48)
The first step is catalyzed by ACOXs that directly interact with molecular oxygen and produce H2O2. H2O2 is subsequently converted to molecular oxygen by catalase. There are at least three ACOXs. ACOX1 preferentially degrades long and medium saturated and unsaturated straight chain fatty acids, whereas ACOX2 has a high affinity for 2-methyl branched fatty acids such as pristanoyl-CoA, DHCA-CoA and THCA-CoA. ACOX3 is also known to be involved in the degradation of 2-methyl branched fatty acids. Unlike the rat homolog, the human gene is expressed in very low amounts in the liver and the physiological role of ACOX3 has yet to be elucidated.
In the second and third steps, L- and D-bifunctional protein (L-PBE and D-PBE) function as enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase, respectively. They catalyze the same set of reactions via mirror image stereochemistry. It has been determined that D-PBE catalyzes the hydration and subsequent dehydrogenation of enoyl-CoA esters of VLCFAs, pristanic acid, DHCA, and THCA. In contrast, the physiological role of L-PBE is not well understood.
In the last step, the two thiolases 3-ketoacyl-CoA-thiolase 1 (pTH1) and sterol carrier protein X (SCPX) (pTH2) are involved in the thiolytic cleavage of 3-ketoacyl-CoA to acetyl-CoA and acyl-CoA shortened by two carbon atoms. pTH1 metabolizes only straight-chain fatty acids. The branched chain fatty acids and bile acid precursors are solely cleaved by pTH2. Once the fatty acid chains are shortened to medium-chain fatty acyl-CoA via peroxisomal β-oxidation, they are conjugated to carnitine, exit the peroxisomes, and undergo further β-oxidation in mitochondria. In contrast, the choloyl-CoA and deoxycholoyl-CoA produced in peroxisomes are converted to taurine- or glycine-conjugated cholic acid, or deoxycholic acid, by bile acid-CoA: amino acid N-acyltransferase and then exported into the cytosol.
Furthermore, peroxisomes catalyze the synthesis of the ether-phospholipids known as plasmalogens, a family of phospholipids in which one of the hydrocarbon chains is joined to glycerol by an ether bond. They are an important membrane constituent in the heart and brain. In addition to lipid metabolism, peroxisomes play a role in several non-lipid metabolic pathways, including purine, polyamine, glyoxylate and D-amino acid metabolism. In addition, peroxisomes are involved in reactive oxygen metabolism. Recently, a new function of peroxisomes was reported by Dixit et al.49) They suggested that peroxisomes as well as mitochondria are important sites of antiviral signal transduction through a sensor of cytosolic virus, called MAVS. Furthermore, Chu et al. reported a novel peroxisomal function in cellular cholesterol transport.50) They demonstrated that peroxisomes have a role in the transport of free cholesterol from lysosomes to the ER through lysosome-peroxisome membrane contact based on genome-wide RNA interference (RNAi) screening. They found that cholesterol transport from the lysosomes is impaired in HeLa cells whose peroxisome genes expression was decreased. It is thus likely that peroxisomes are multifunctional organelles that interact with other organelles. Crosstalk of peroxisomes with mitochondria, lysosome and other organelles is important in various metabolic and signaling pathways.
3. THE ABC TRANSPORTER SUBFAMILY D
3.1. General Characteristics of ABC TransportersThe ABC transporters comprise one of the membrane-bound protein superfamilies in prokaryotes and eukaryotes. The structure of the ABC transporters is highly conserved, and they catalyze ATP-dependent transmembrane transport of a variety of substrates into or out of cells so as to maintain cellular homeostasis. The human ABC transporter family is currently known to comprise 48 members, if we exclude ABCC13, which is a pseudogene product without any functional ABC protein domain. The ABC transporters are divided into seven subfamilies, A–G, based on structural organization and amino acid homology.51,52) The human ABC proteins are involved in a number of important physiological processes, and defects in their function have been shown to be responsible for various diseases such as Tangier disease (ABCA1) and X-linked adrenoleukodystrophy (X-ALD) (ABCD1).
3.2. ABCD TransportersFour ABCD transporters have been identified to date: adrenoleukodystrophy protein (ALDP/ABCD1), ALDP-related protein (ALDRP/ABCD2), the 70-kDa peroxisomal membrane protein (PMP70/ABCD3), and the PMP70-related protein (P70R/ABCD4).53–58) The ABCD3 gene was first cloned as a peroxisomal ABC transporter from rat liver cDNA library by Kamijo in the Hashimoto lab at Shinshu University. The predicted amino acid sequence revealed that the COOH-terminal region in ABCD3 has strong sequence similarities to a group of ABC transporters that includes MalK and Mdr. ABCD3 is markedly induced by administration of hypolipidemic agents in parallel with peroxisome proliferation and induction of peroxisomal β-oxidation enzymes.53) This was the first report of an ABC transporter on intracellular organelles in eukaryotic cells.
Then, the ABCD1 gene was identified by positional cloning that was performed by Aubourg’ group at INSERM (France) to identify the gene responsible for X-ALD.54) The deduced protein sequence showed significant sequence identity with ABCD3. There was considerable surprise at this unexpected result since it was suspected at that time that VLCFA-CoA synthetase was the gene responsible for X-ALD. The ABCD2 gene was cloned from a cDNA library from the mouse monocyte/macrophage cell line P388D1 using PCR products encoding Walker A and a 12-mer conserved motif in the ABC transporters.55) ABCD4 was identified by a homology search for ABCD1 and ABCD3 related sequences in a database of expressed sequence tags (ESTs).57,58)
ABCD1–3 are known as peroxisomal proteins and are involved in the transport of various kinds of fatty acids. ABCD4 localizes to lysosomes and is suggested to be involved in the transport of vitamin B12 from the lysosome to cytosol (see below).
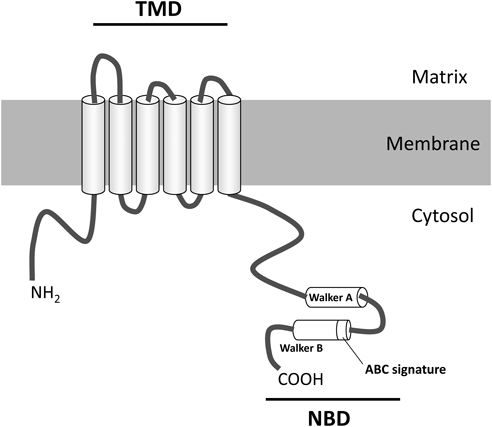
ABCD transporters are predicted to be ABC half-transporters structure, with one transmembrane domain (TMD) and one nucleotide-binding domain (NBD). From the hydropathy profile, the amino terminal half is hydrophobic, with six transmembrane segments, and the COOH-terminal half is hydrophilic, having NBD (Fig. 5). TMD provides the passageway for the substrate across the membranes and NBD energizes the directional transport of these substrates by alternating cycles of ATP binding and hydrolysis. NBD contains three conserved motifs: Walker A, Walker B and ABC signature motif, which is situated upstream of the Walker B sequence.51,59) The NBD in ABCD transporters is suggested to be exposed to the cytosol and this was confirmed by protease treatment of isolated peroxisomes.60)
The ABC proteins transport substrate through the conformation change induced by the cycle of ATP binding and hydrolysis. We showed that the binding and hydrolysis of ATP induced conformational changes in ABCD3 close to the boundary between the TMD and NBD, and the helical domain between the Walker A and B motifs.61,62) On the other hand, Guimarães et al. proposed that the TMD of ABCD1 is involved in the recognition of substrates such as long- and VLCFA-CoA from evidence that the substrate induced conformational alterations in ABCD1 with a protease-based assay.63) These findings suggest that the NH2-terminal TMD of ABCD1 is involved in recognition of these substrates, and undergoes a conformational change on ATP binding to the COOH-terminal NBD of ABCD1. Recently, we expressed ABCD1–4 in the methylotrophic yeast Pichia pastoris and reconstituted ABCD1–4 into liposomes after purification of these proteins. ABCD1–4 displayed stable ATPase activity.64) These liposomes appear useful for characterizing the mechanism by which substrate is transported.
In terms of the quaternary structures of the peroxisomal ABC proteins, ABCD1 and ABCD3 are mainly found as a homodimer in mammalian peroxisomal membranes,65–67) although ABCD1 is able to form a complex with ABCD2 or ABCD3. Recently it was reported that ABCD1 and ABCD2 might assemble as a heterotetramer.68)
3.3. Targeting of ABCD TransporterAn NH2-terminal hydrophilic region containing an H0 motif plays an important role in trafficking of ABCD transporters.69) The H0 motif is a hydrophobic segment adjacent to the NH2-terminal portion of TMD1. ABCD transporters are translated on free polysomes. ABCD1–3 possessing the H0 motif are selectively captured by Pex19p, which is essential for the early steps of peroxisome biogenesis and most likely also involved in peroxisomal membrane synthesis then destined for peroxisomes.70) In contrast, ABCD4 hardly interacts with Pex19p because it lacks the NH2-terminal H0 motif, and as a result, ABCD4 is recognized by signal recognition particles (SRPs) and integrated into the ER membrane.71) Subsequently, ABCD4 is translocated to lysosomes72) (Fig. 6). In the GTOP database (http://spock.genes.nig.ac.jp/~genome/gtop.html), 193 organisms have ABCD proteins with homology to mammalian, yeast, or plant ABCD proteins. We performed Kyte–Doolittle hydropathy plot analysis on the known ABCD proteins and found that at least 60 of 143 organisms appeared to have ABCD proteins lacking the typical NH2-terminal H0 motif. Caenorhabditis elegans pmp-5, Arabidopsis thaliana ABCD2, and Mucor circinelloides ABCD protein MC91590 lacking H0 motif were targeted to the ER but not peroxisomes.69) ABC transporters lacking the H0 motif might be functional in the ER or lysosomes.
In terms of the trafficking of the newly synthesized peroxisomal ABC transporters, ABCD3 has been characterized in some detail.73–76) Because ABCD3 is a hydrophobic integral membrane protein, binding with Pex19p is indispensable for ABCD3 to retain proper conformation as a soluble form for targeting to peroxisomes. To investigate the critical region of ABCD3 (AA.1–659) for targeting to peroxisomes, we prepared various forms of NH2-terminal or COOH-terminal truncated ABCD3 fused to GFP.75) The COOH-terminally truncated ABCD3 (AA.1–144)-GFP, comprising TMD1 and TMD2, along with GFP-ABCD3 (AA.263–375), containing TMD5 and TMD6, were found to be localized to peroxisomes. Further analysis using mutated ABCD3 revealed that ABCD3 is recognized and binds to Pex19p at the NH2-terminal hydrophobic motif constituted by Leu21-Leu22-Leu23 and the region of TMD5–TMD6. Subsequently, using another version of mutant ABCD3 by site-directed mutagenesis, we suggested that the hydrophobic amino Ile70-Leu71 and Ile307-Leu308 adjacent to TMD1 and TMD5, respectively, are essential for targeting to peroxisomes as mPTS. Thus ABCD3 forms a complex with Pex19p that is transported to peroxisomes by mPTSs. Finally, ABCD3 is inserted into the peroxisomal membranes through a putative proteinaceous receptor on the peroxisomal membrane. In this process, at least two TMDs (TMD1 and TMD2) are required for proper insertion75) (Fig. 7).
The targeting of ABCD1 to peroxisomes has also been characterized.77,78) It was determined that a 14-amino acid motif adjacent to the deduced TMD1 (F(F/L)X(R/Q/K)(L/F)(L/I/K)XLLKIL(F/I/V)P/) functions as an mPTS of ABCD1, and it was demonstrated that substitution or deletion of these hydrophobic residues significantly reduced targeting efficiency. In particular, deletion of the three amino acids Leu78-Leu79-Arg80 was critical for the peroxisomal targeting of ABCD1. This region corresponds to Ile70-Leu71-Lys72 in ABCD3, which is the mPTS in ABCD3. In ABCD2, a potential Pex19p binding site was also identified as ABCD1. It corresponds to AA.84–97, which are localized in proximity to the putative TMD1. However, no experimental data are available as yet to support the functionality of this putative Pex19p binding site as well as mPTS.
Newly synthesized ABCD4 is inserted into ER membranes and then translocated to lysosomes through interaction with the lysosomal membrane protein LMBD1. Recently, we demonstrated the underlying mechanism of this translocation.72) When ABCD4 was expressed alone in HuH7 cells, ABCD4 was localized to ER membrane as a homodimer. However, coexpression of LMBD1 in the cells drastically changed the localization of ABCD4 to lysosomes. During this translocation, the ABCD4 dimer formed a complex with LMBD1. Concerning the targeting of LMBD1 to lysosomes, a putative AP-2 binding motif, Tyr233-Glu234-Arg235-Leu236 is essential.79) When this mutant LMBD1 and ABCD4 were coexpressed, the distribution pattern of ABCD4 was superimposable on the mutant LMBD1 on the plasma membrane of the cells. Furthermore, it was confirmed by equilibrium iodixanol density gradient centrifugation that endogenous ABCD4 localizes to lysosomes and ER in HEK293 cells. Lysosomal localization of ABCD4 was reduced to approximately 40% in LMBD1 knockout HEK293 cells. These results show that LMBD1 is responsible for the localization of endogenous ABCD4. Subcellular localization of ABCD4 is determined through its association with adaptor proteins.
In terms of the localization of ABCD transporters to the organelles, the NH2-terminal amino acid sequence before TMD1 suppresses targeting of the proteins to the ER. The NH2-terminal amino acid sequence (AA.1–80) of ABCD3 is transported to mitochondria, although the H0 motif is included in the sequence. However, the sequence with TMD1 and 2 (AA. 1–144) targets peroxisomes.80) The evidence suggests that during the translation of ABCD3 it escapes association with SRP by an interaction with HPS70, which is required for targeting to mitochondria. After translation of the TMD1–2 sequences, ABCD3 can shift the binding of HPS70 to Pex19p and the complex targeting to the peroxisome. When comparing the surface area of peroxisomes with that of mitochondria and the ER, the peroxisomal area is considerably less. Therefore, living organisms may have adapted correct PMP targeting to the peroxisome without any mistargeting to mitochondria and ER membranes by the using the amino acid sequence of H0 together with TMD1 and 2.
3.4. Functions of ABCD TransportersABCD1 and ABCD2 have a high degree of sequence homology, and both are involved in the transport of saturated and unsaturated VLCFA-CoA into peroxisomes81,82) (Fig. 8). ABCD3 plays a role in the import of bile acid intermediates, as well as pristanic acid and phytanic acid, because Abcd3-deficient mice show impaired metabolism of these fatty acids.83) ABCD4 might be involved in the export of vitamin B12 from lysosomes into cytosol.84) Concerning the transport of mammalian ABC transporters, almost all function as exporters that transport substrate from the cytosol to the exterior of the cell or the lumen of subcellular organelles, which is considered to be an “extracellular space” in eukaryotic cells. At present, ABCA4 is reportedly the only example of a mammalian ABC importer that actively flips N-retinylidene-phosphatidylethanolamine from the lumen to the cytoplasmic leaflet of the disc membrane in retinal photoreceptor cells.85) If ABCD4 actually transports vitamin B12 as we postulate, it would be the first mammalian ABC importer shown to transport a soluble compound.
3.4.1. ABCD1It is thought that ABCD1 functions as a transporter of VLCFA across the peroxisomal membrane for β-oxidation. This is supported by the result that exogenous expression ABCD1 into X-ALD skin fibroblasts restored VLCFA β-oxidation, and consequently, the VLCFA content decreased to normal in the fibroblasts.86,87) Furthermore, it was demonstrated that ABCD1 is involved in the transport of saturated, monounsaturated and polyunsaturated VLCFA-CoA across peroxisomal membranes by expressing human ABCD1 in a pxa1/pxa2 knockout yeast mutant.81) However, the mechanism of fatty acid transport by ABCD1 has not been well characterized. It is proposed that the CoA moiety of VLCFA-CoA is cleaved during the transport cycle since Comatose, a homolog of human ABCD1 in A. thaliana, itself possesses intrinsic acyl-CoA thioesterase activity.88) In addition, β-oxidation of oleic acid was analyzed using yeast cells with a deficiency of thiolase and expressing carnitine palmitoyltransferase 2-SKL in peroxisomes in medium, including [18O]H2O.89) It is suggested that acyl-CoA is hydrolyzed before being metabolized by quantitation of the amount of isotope-labeled 3-OH-C18-1-carnitine. Another model suggests that esterified fatty acids are delivered directly to the peroxisomal matrix. This model depends on the observation that the β-oxidation of VLCFA-CoA in peroxisomes isolated from fibroblasts directly depends on ABCD1 without a need for additional re-esterification by an acyl-CoA synthetase.90)
Recently we used proteoliposomes reconstituted with purified ABCD1 to show that that ABCD1 expressed in the methylotrophic yeast possesses acyl-CoA thioesterase.64) It is interesting how acyl-CoA thioesterase activity involves transport of VLCFA into peroxisomes. The activity of ABCD1 was very low compared with other acyl-CoA thioesterase found in peroxisomes, mitochondria, the ER and cytosol in mammals.91–95) In addition, the acyl chain covalently bound to ABCD1 was shown to be detectable under a condition in which such intermediates were not detectable in other forms of acyl-CoA thioesterase. This suggests that acylation of ABCD1 occurs over a relatively long period of time. Therefore, acyl-CoA thioesterase might be required to regulate transport of VLCFA through acylation of the ABCD1 protein.
Export of cholesterol from lysosomes is reduced by the mutation of ABCD1.50) Although the mechanism is not clear at present, it is interesting that there is crosstalk between the peroxisome and lysosome in cholesterol metabolism. ABCD1 gene expression is reportedly affected by the cholesterol levels in human THP-1 cells and primary macrophages.90) These findings suggest a link between ABCD1 function and cellular cholesterol metabolism.
3.4.2. ABCD2ABCD2 displays functional redundancy with ABCD1.96) However, the phenotype of fatty acid abnormalities in Abcd2−/− mice is different from that in Abcd1−/− mice.97)Abcd1−/− mice display a higher accumulation of C24:0 and especially C26:0. In contrast, Abcd2−/− mice display fatty acid abnormalities, especially monounsaturated and polyunsaturated fatty acids, indicating different substrate specificities of ABCD1 and ABCD2. This is supported by experiments using the yeast pxa1/pxa2Δ mutant, which expresses ABCD1 and/or ABCD2.81) ABCD2 shows overlapping substrate specificities with ABCD1 toward saturated and monounsaturated fatty acids. ABCD1 has a higher specificity for saturated VLCFA-CoA. In contrast, ABCD2 but not ABCD1 has an affinity for polyunsaturated fatty acids such as C22:6-CoA and C24:6-CoA (Fig. 8). The β-oxidation activity of C24:6n-3, an immediate precursor of docosahexaenoic acid (DHA), is reduced in Abcd2-deficient mice brain. In addition, ABCD2 expression is highly sensitive to dietary polyunsaturated fatty acids (PUFAs), suggesting that Abcd2 is involved in the transport of PUFAs in relation to DHA metabolism.81,97,98) Furthermore, the Abcd2 expression pattern in tissues and cells in mice and humans is different from that of ABCD1. It is hypothesized that there are benefits for cells by the expression of either ABCD1 or ABCD2.
ABCD2 appears to have a central role in the metabolism of monounsaturated and polyunsaturated VLCFA rather than saturated VLCFAs, and may be involved in the regulation of oxidative stress and synthesis of DHA.99) However, the actual function of ABCD2 in vivo is still unclear because the endogenous expression level of ABCD2 is quite low in human cells and there is no reported disease caused by ABCD2 dysfunction.
3.4.3. ABCD3ABCD3 is one of the most abundant PMPs, at least in the liver and kidney, and has been reported to be involved in transport of various fatty acids (Fig. 8). We demonstrated that overexpression of ABCD3 stimulates C16:0 β-oxidation activity in Chinese hamster ovary (CHO) cells, but inhibits C24:0 β-oxidation activity.100) In addition, we showed that C24:0 β-oxidation activity was not decreased by silencing of ABCD3 in ABCD1-knockdown U87 cells,101) suggesting that ABCD3 does not contribute to peroxisomal VLCFA β-oxidation. ABCD3 has been shown to have a role in the transport of more hydrophilic substrates compared to VLCFA. It has been reported in Abcd3-deficient mice that the bile acid precursors THCA and DHCA, as well as pristanic acid, accumulate in plasma. There was a significant reduction of chenodeoxycholic acid and cholic acid conjugated with taurine or glycine in the liver, bile, and intestine. On the other hand, a remarkable increase of bile acid intermediates with C27 was observed. There was a marked accumulation of phytanic and pristanic acid in the plasma of Abcd3−/− mice when supplied a phytol diet.83) These results suggest that ABCD3 is active in the transport of LCFA-CoA, THCA-CoA, DHCA-CoA, and branched chain acyl-CoA (Fig. 8). In the mouse liver, ABCD3 expression is induced by treatment with fibrates, such as ciprofibrate and fenofibrate, via PPARα. Recently, a patient with ABCD3 defect has been identified.83) The patient exhibited hepatosplenomegaly and severe liver disease as well as a significant accumulation of DHCA and THCA in plasma. ABCD3 possesses intrinsic acyl-CoA thioesterase activity, as do ABCD1 and ABCD2.64)
3.4.4. ABCD4Recently, new patients were discovered with a vitamin B12 (cobalamin) deficiency associated with mutation of ABCD4, suggesting that ABCD4 is involved in vitamin B12 metabolism.84) In humans, cobalamin forms a complex with transcobalamin in the blood stream. This complex is endocytosed into cells and delivered to lysosomes. Cobalamin in the lysosomes is then exported to the cytosol and in part to mitochondria, and converted into the two active cofactors methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl).102) A defect in ABCD4 results in accumulation of cobalamin in lysosomes. In patient fibroblasts, intracellular enzyme-bound cobalamin level is significantly lower than in control fibroblasts. Expression of wild-type ABCD4 in patient fibroblasts was shown to normalize intracellular enzyme-bound cobalamin levels and remarkably increase the synthesis of MeCbl and AdoCbl. Expression of mutant ABCD4 at the putative ATP-binding site leads to reduced synthesis of both cofactors. This suggests that ATPase activity of ABCD4 may be involved in the intracellular processing of cobalamin. As mentioned above, mutations in ABCD4 and LMBD1 result in a quite similar phenotype.103) This finding suggests that these two proteins function as a complex in the process of transporting cobalamin from lysosomes to the cytosol. The bacterial ABC transporter BtuCD is involved in the import of cobalamin into the cytoplasm across the inner membrane and possesses ATPase activity.104) ABCC1, a multidrug resistance-associated protein on the plasma membranes of mammalian cells, may be involved in the cobalamin export from cells.105) ABCC1 itself also possesses ATPase activity. Therefore, we postulate that ABCD4 comprises a transporter unit and that LMBD1 is an accessory protein. LMBD1 is a highly glycosylated protein and this might protect ABCD4 from proteases in lysosomes.
ABCD1–3 transport substrates from the cytosol to peroxisomes. In contrast, ABCD4 reportedly transports cobalamin from lysosomes to the cytosol, although this has not yet been demonstrated experimentally. It is of special interest to determine the mechanism underlying the difference in the direction of this transport.
4. PEROXISOME DISEASE AND X-LINKED ADRENOLEUKODYSTROPHY
Peroxisomes are indispensable organelles in humans and have many metabolic functions. Peroxisome disease is characterized by the accumulation of non-degraded molecules such as VLCFA, THCA, DHCA, and phytanic acid and reduction of certain physiologically essential molecules such as DHA, bile acids, and ether-phospholipids. Peroxisome diseases are congenital metabolic disorders involving mutations of genes encoding peroxisomal proteins and/or proteins involved in peroxisome biogenesis. These diseases are classified into two groups: PBD and single peroxisomal enzyme deficiency.106)
4.1. Peroxisome Biogenesis DiseaseBiogenesis of peroxisomes including import of PMPs and peroxisomal matrix proteins is mediated by many peroxins (Fig. 2). PBD is a genetically heterogeneous group of autosomal recessively inherited disorders with a specific defect in one of the 13 different PEX genes. The majority of PBD are caused by biallelic mutations. Recently, patients with mutations in the genes DLD/DRP1 and PEX11β, which are involved in peroxisome fission, were identified.107–109)
Clinically, PBD ranges from severe, frequently lethal disorders to milder, late-onset progressive neurological disease. The patients show severe hypotonia of muscles, hepatomegaly with liver dysfunctions, variable neurodevelopmental delay, retinopathy and perceptive deafness. Disease severity is dependent on the role of proteins involved in peroxisome biogenesis and the degree of residual function that exists in mutant proteins. The most severe examples are Zellweger syndrome, neonatal adrenoleukodystrophy (which is different from X-linked adrenoleukodystrophy, X-ALD) and infantile Refsum disease. The diseases are classified based on the severity of the clinical symptoms. The following mutations cause these diseases. Mutation of the PEX3, PEX16, and PEX19 genes impacts the biogenesis of peroxisomal membranes through PMP import. In contrast, mutation of the PEX1, PEX2, PEX5, PEX6, PEX10, PEX12, PEX13, PEX14, and PEX26 genes, respectively, affects the import of matrix proteins into peroxisomes. In the case of PEX7 mutation, only the import of peroxisomal matrix proteins with PTS2 is affected. The mutation causes rhizomelic chondrodysplasia punctate (RCDP) type 1. These patients exhibit limited peroxisomal metabolic abnormalities such as deficiency of ether-phospholipid synthesis and α-oxidation of phytanic acid. The phenotype of the disease is milder than Zellweger syndrome. RCDP is a clinical phenotype, and five genetically distinct subtypes have been reported to date. Type 1 is PBD, while types 2, 3, and 4 are single peroxisomal enzyme deficiencies (see below). Recently, type 5 was found to be caused by mutation of Pex5pL. Mutations of DLP/DRP1 and PEX11β were shown to cause a novel class of peroxisomal fission disorders.107,108) In addition, TRIM37 ubiquitylates Pex5p on peroxisomal membranes. TRIM37-mediated ubiquitylation stabilizes Pex5p and promotes peroxisomal matrix protein transport. It has been shown that TRIM37 gene mutations cause muscle-liver-brain-eye (mulibrey) nanism.110)
4.2. Single Enzyme DeficiencyThe diseases due to single peroxisomal enzyme deficiency include defects of peroxisomal matrix enzymes as well as PMPs. The clinical phenotypes of single peroxisomal enzyme deficiency thus depend on the specific function of a given protein in peroxisomal metabolism. Single enzyme deficiencies in peroxisomes induce biochemical abnormalities that frequently result in severe disease. Mutation of ABCD1 causes X-ALD. ABCD3 deficiency has been reported to cause persistent hepatosplenomegaly and severe liver disease.83) Deficiency of β-oxidation enzymes causes acyl-CoA oxidase 1 deficiency, D-bifunctional protein (multifunctional protein 2; MFP2) deficiency, 2-methylacyl-CoA racemase deficiency, and SCPx deficiency. In addition, acyl-CoA oxidase 2 deficiency causes impaired bile acid synthesis since it oxidizes branched-chain acyl-CoA. Bile acid-CoA: amino acid N-acyltransferase deficiency causes intrahepatic cholestasis since bile acid amidation is necessary for export of bile acid to cytoplasm. Deficiency of the α-oxidation enzyme phytanoyl-CoA hydroxylase results in Refsum disease. Dysfunction of enzymes involved in ether-phopholipid synthesis such as DHAPAT, alkyl-DHAP synthetase, and fatty acyl-CoA reductase causes RCDP types 2, 3, and 4, respectively. Dysfunction of glyoxylate detoxification by mutation of alanine: glyoxylate aminotransferase and deficiency of catalase involved in H2O2-metabolism causes hyperoxaluria type I and acatalasemia, respectively. Contiguous ABCD1 DXS1357 deletion syndrome (CADDS) has a deletion that extended into the promoter region of ABCD1 and neighboring gene DXS1357E/BCAP31. The phenotype is similar to Zellweger syndrome. Recently, mutation of the acyl-CoA binding domain containing protein 5 (ACBD5) that is located on peroxisomal membranes was shown to result in impaired VLCFA metabolism, leading to retinal dystrophy and white matter disease.111,112) It is suggested that ACBD5 interacts with VAMP-associated proteins A and B on the ER, and that this interaction might be required to facilitate lipid exchange between peroxisomes and the ER.113)
Peroxisomal diseases are rare and severe conditions. The development of effective treatments is one of the most important challenges in the field. In the case of X-ALD, the number of patients is relatively large, and analysis of the pathogenesis and development of diagnostic and therapeutic approaches are in progress.
4.3. X-Linked Adrenoleukodystrophy (X-ALD)X-ALD is the most common peroxisomal disease caused by mutations in the ABCD1 gene,54) affecting approximately 1 in 15000–30000 males in the U.S.A. and Japan.114,115) Patients display progressive demyelination in the central nervous system (CNS), adrenal insufficiency, and testicular dysfunction as pathological characteristics. There are several X-ALD phenotypes that are categorized based on the onset and severity of the disease: the childhood cerebral form (CCALD), adolescent cerebral form (AdoCALD), adult cerebral form (ACALD), adrenomyeloneuropathy (AMN), and Addison’s disease alone. The most frequent presentations are CCALD and AMN. The CCALD form is associated with inflammation of the cerebral white matter, cerebral demyelination and adrenocortical insufficiency, with the onset occurring in childhood usually between the ages of 3 and 10 years. In contrast, AMN is characterized by a slow progressive axonopathy in males in an approximate in men aged 20–30 years.
According to the literature,116,117) in 1897 Heubner first described a young boy with rapidly progressive neurologic deterioration consistent with X-ALD who had “diffuse sclerosis” on autopsy. Similar pathological features were described by Ceni in 1899 and by Haberfield and Spieler in 1910. Surprisingly, Schilder’s postmortem examination of the brain in 1913 revealed severe alteration of the white matter with demyelinated lesions combined with an infiltration of lymphocytes and glial cells, signs of an extensive inflammatory response. Siemerling and Creutzfeldt first documented progressive demyelination with atrophy of the adrenal cortex in 1923.118) In 1952, the disease was placed in the group of demyelinating disorders. In 1963, ALD was speculated an X-linked inherited disease119) and in 1970, the name adrenoleukodystrophy was proposed and ultimately came to be known as X-ALD.120) X-ALD was hypothesized a lipid storage disease by the finding that lipid inclusion in several different types of cells, including brain macrophages. In 1976, Igarashi established the biochemical basis for X-ALD by showing accumulation of VLCFA in the brain and adrenal glands of patients.121) In 1993, Aubourg and colleagues identified the ALD gene by positional cloning as the gene in which mutations are responsible for X-ALD.54) The ALD gene is approximately 21 kb long and contains 10 exons encoding an ABC transporter composed of 745 amino acids. Because the VLCFA-CoA synthetase gene had been suspected to be associated with X-ALD at that time, it was surprising that the responsible protein was this transporter.
4.4. Biochemical Features and Pathogenesis of X-ALDMutations in the ALD/ABCD1 gene result in increased VLCFA, not only in the CNS, but also most tissues, plasma, erythrocytes and white blood cells in patients with X-ALD. VLCFA constitutes the ester bonds of phospholipids, gangliosides, and cholesterol esters. In contrast, the amount of polyunsaturated fatty acids such as DHC C22:6 is not elevated. A marked increase of VLCFA (C26:0, C24:0) in plasma is one of the best diagnostic markers for X-ALD. Recently, it was pointed out that the amount of 1-hexacosanoyl-2-lyso-sn-3-glycerophosphorylcholine (C26:0-lyso-PC) in whole blood is a potent diagnostic marker and measurement of the lipid content in a dry blood spot is expected to help in neonatal screening for X-ALD.122)
In X-ALD, brain glial cells and adrenal cortex predominantly expressing ABCD1 are targets for the disease. The pathogenic mechanism of neurodegeneration, especially demyelination, in X-ALD remains obscure. In CNS, increase of phospholipids with VLCFA in astrocytes and microglial cells might cause mitochondrial dysfunction. Subsequently, proinflammatory cytokines are released from astrocytes and microglial cells through oxidative stress and dysregulation of innate immune response, resulting in the demyelination123,124) (Fig. 9). Evidence based on in vitro and in vivo studies suggests that oxidative stress contributes to the pathogenesis of the cerebral inflammatory phenotype.125) Proinflammatory cytokines are known to have negative effects on oligodendrocytes. Bone marrow transplantation may prevent progression of the demyelination that takes place.
A characteristic feature of X-ALD is that there are large differences in the onset and progressive course of the disease among patients, even those who possess the same mutation in the same ABCD1 gene, suggesting mutation of other genes and environment factors together with mutation of the ABCD1 gene might be involved in the development of X-ALD.126) Therefore, it is important to identify such modifiers in the development of the disease.
4.5. Therapeutic ApproachesAt present, there is no effective treatment to prevent the onset or the progression of neurological pathogenesis in X-ALD. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only currently available therapeutic procedure that can halt cerebral demyelination in patients with early stages of CCALD.127) The mechanism by which HSCT stops demyelination has not yet been made clear. It seems likely that HSCT allows stem cells to penetrate into the brain parenchyma through the brain–blood barrier and the engrafted stem cells develop into microglia-like cells. However, HSCT is still associated with a high risk of mortality because of graft-versus-host reaction and immunodeficiency induced by myeloablation. Dietary therapy with Lorenzo’s oil is now a commonly accepted treatment for X-ALD patients. However, Lorenzo’s oil does not halt the progression of the cerebral X-ALD phenotype in patients who were already symptomatic when the treatment was initiated.128)
In addition to HSCT-based therapy, novel pharmacological therapies may delay the onset and progression of the disease. Many candidate compounds have been reported for the treatment of X-ALD to date.117,129) Concerning the development of therapeutic compounds, the following approaches are underway or soon expected (Fig. 10): 1. Stabilization and correct subcellular localization of missense ABCD1 mutants with residual activity might be an effective way to recover the dysfunction of ABCD1. 2. Induction of ABCD2 expression. 3. Stimulation of peroxisomal fatty acid β-oxidation. 4. Inhibition of microsomal fatty acid elongation. 5. Attenuation of the dysregulated response to oxidative stress has as yet untapped potential as an effective approach in X-ALD. Chemical compounds that can pass through the blood–brain barrier and act on these targets would be promising drugs for X-ALD therapy, since the target cells in X-ALD are astrocytes and microglial cells in the CNS.
More than 70% of the patient fibroblasts with a missense mutation display either a lack or reduction of the ABCD1 protein because of post-translational degradation. We analyzed the stability of the missense mutant ABCD1 proteins in X-ALD fibroblasts and found that some of the mutant ABCD1 proteins have capacity to recover function by incubation at a low temperature.130,131) In the case of such mutations, chemical compounds that stabilize mutant ABCD1 proteins are therapeutic candidates. We prepared CHO cell lines stably expressing ABCD1 proteins with a missense mutation in fusion with GFP at the COOH-terminal. The stability of each mutant ABCD1-GFP in CHO cells was similar to the corresponding mutant ABCD1 protein in X-ALD fibroblasts. These findings prompted us to use CHO cells expressing mutant ABCD1-GFP for a screening of chemical compounds that can stabilize the mutant ABCD1 protein. We established a fluorescence-based assay method for the screening of chemical libraries.131) We screened 1948 compounds from a library of existing drugs and identified bortezomib, a drug for multiple myeloma. Because bortezomib exerts severe side effects, it is obviously not applicable for X-ALD patients at present. Instead, bortezomib provides a piece of supporting evidence that stabilization of a subset of mutant ABCD1 proteins could have therapeutic value for X-ALD in the future.
Accumulation of VLCFA in the CNS may be a major culprit in X-ALD pathogenesis. It is known that approximately 30–40% of peroxisomal fatty acid β-oxidation activity still remains in X-ALD patient fibroblasts. Therefore, chemical compounds that stimulate the residual peroxisomal fatty acid β-oxidation, including peroxisomal function, are expected to be lead compounds in X-ALD treatments.132) We found that the fluorescent fatty acid analog 12-(1-pyrene)dodecanoic acid (pyrene-C12:0) acts as a substrate for peroxisomal fatty acid β-oxidation, not mitochondrial fatty acid β-oxidation, and the products are detectable by reverse phase (RP)-HPLC. In X-ALD fibroblasts, β-oxidation activity for pyrene-C12:0 was found to be approximately 40% of that in control fibroblasts, which is consistent with previous results using [1-14C]lignoceric acid as substrate. We established a convenient procedure for the screening of chemical compounds.133) Cells were incubated in a medium containing pyrene-12 : 0. Thereafter, the lipid fractions were extracted and subjected to alkaline hydrolysis to liberate the fatty acids. The fluorescent peaks correspond to the shortened pyrene-fatty acids separated by RP-HPLC. We currently use this procedure and a screening of compounds is in progress.
4.6. New Approaches to X-ALDAutologous HSCT-gene therapy, in which the ABCD1 gene is transduced into hematopoietic stem cells by a lentivirus vector, has been tested has been tested and found advantageous in CCALD who do not have any HLA-matched donors. In 2009, Cartier et al. reported that lentiviral-mediated gene therapy of hematopoietic stem cells halted the progression of X-ALD.134) Demyelination in the brain was not observed by magnetic resonance imaging (MRI) scan for 3 years after the transplantation. These results strongly suggest that hematopoietic stem cells were transduced in the patients. Beginning 14–16 months after infusion of the genetically corrected cells, progressive cerebral demyelination stopped in the two patients, a clinical outcome comparable to that achieved by allogeneic HSCT. Thus, lentiviral-mediated gene therapy of hematopoietic stem cells evidently provides clinical benefit in X-ALD. Recently, 17 cases received with gene therapy and positive results were obtained except for one case.135) These successful cases are epoch-making. However, at present gene therapy is limited to patients at an early stage close to onset. The mechanism by which HSCT stops demyelination has yet to be elucidated. It seems likely that HSCT allows stem cells to penetrate into the brain parenchyma through the brain blood barrier, where the engrafted stem cells act as microglia-like cells. We showed that transplanted wild type stem cells to ABCD1 knockout mice penetrated into the brain and differentiated into microglial cells, not astrocytes (unpublished observation).
Age at onset, development of the disease, and degree of severity vary widely among X-ALD patients. HSCT at an early phase of the disease is the only effective therapeutic approach to X-ALD at present. Thus, the discovery of biomarkers for the early detection or even the prediction of the onset of X-ALD is urgently needed. The diagnosis of X-ALD is based on the C26:0/C22:0 ratio determined by fatty acid composition in plasma. However, some additional VLCFA-containing lipid species may contribute to symptom onset and wide variation in the X-ALD phenotype. Lipidomic research involves identification and quantification of thousands of molecular lipid species and their interactions with additional lipids, proteins. and other metabolites. Recent progress in LC/MS has made it possible to perform a metabolome. LC-electrospray ionization-MS/MS (LC-ESI-MS/MS) has contributed to the determination of individual phospholipid species. Investigation of the relations between the diversity of VLCFA-containing lipids and the pathogenesis of peroxisomal diseases is underway.136–138) It appears that phospholipids with VLCFA, with or without double bonds, might exert effects on cellular functions. The molecular mechanism for the incorporation of saturated or monounsaturated VLCFA into the sn-1 position of phospholipids is involved in the distribution of phospholipids in the cells, and how phospholipids are distributed will be key to understanding the development of X-ALD.
A recent induced pluripotent stem (iPS) cell method has opened new directions in understanding the pathogenesis of many intractable diseases, allowing disease models and cell replacement therapy. Jang et al. established iPS cells from X-ALD patients and demonstrated that oligodendrocytes differentiated from X-ALD-induced iPS cells displayed an accumulation of C26:0. Interestingly, C26:0 accumulation was reported to be much higher in CCALD oligodendrocytes than AMN oligodendrocytes. In addition, several studies on the pathogenesis of X-ALD have been reported.139–141)
Exome is the portion of the genome that is formed by exons. It consists of all of the DNA that is transcribed into mature RNA in cells. The exome of the human genome comprises roughly 180000 exons constituting about 1% of the total genome. Mutations in the exome are thought to harbor 85% of those that exert an effect on disease. Exome sequencing has proved to be an efficient strategy for determining the genetic basis of diseases. Recently, new gene mutations have been identified in exome sequences in peroxisomal diseases.142,143)
5. Concluding Remarks
Since 1985, I have intensively studied the biogenesis and function of peroxisomes, with a particular focus on ABC transporters and X-ALD. Here I have looked back on this work and reviewed the biogenesis and function of peroxisomes, which together have been the focus of our studies. According to the development of science and technology, our understanding about biogenesis and function of peroxisome has deepened. Generally, when we begin to understand a mechanism, additional questions emerge and we would like to know more precisely exactly how the mechanism operates. That is science. It is to be hoped that a better understanding of peroxisomes will flow from new evidence obtained by ever more sophisticated new experiments. One of the outstanding unresolved questions is where pre-peroxisomes are formed. In addition, it is important to elucidate the molecular mechanism underlying crosstalk of peroxisomes with mitochondria and other organelles in various metabolic signaling pathways. This crosstalk is suggested to be important in the regulation of cellular function through exchange of metabolites and signaling molecules as well as direct contact.
With regard to peroxisome diseases, many genes have been identified and understanding of their pathogenesis is a work in progress. Many of these diseases are severe so progress is urgently required. At present, only X-ALD is successfully targeted. I hope in the near future that progress in X-ALD will serve as a model approach to other peroxisome diseases. This review was written based on the article, Imanaka T. Yakugaku Zasshi 2018; 138: 1067–1083 (review, Japanese).
Acknowledgments
I would like to express sincere gratitude to all the members including the graduate and undergraduate students of my research groups in Teikyo University and University of Toyama. I also express cordial thanks to all the collaborators in many universities and companies. It is my pleasure that I was able to participate in research on peroxisomes with so many people. This research was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science and for Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan. Dr. Kevin Boru reviewed the manuscript prior to submission.
Conflict of Interest
The author declares no conflict of interest.
Note
This review of the author’s work was written by the author upon receiving the 2016 Pharmaceutical Society of Japan Award for Divisional Scientific Contribution.
REFERENCES
- 1) Rhodin J. Correlation of ultrastructural organization and function in normal and experimentally changed proximal convoluted tubule cells of the mouse kidney. Ph. D. thesis. (1954).
- 2) de Duve C. Microbodies in the living cell. Sci. Am., 248, 74–84 (1983).
- 3) de Duve C, Baudhuin P. Peroxisomes (microbodies and related particles). Physiol. Rev., 46, 323–357 (1966).
- 4) Leighton F, Poole B, Beaufay H, Baudhuin P, Coffey JW, Fowler S, De Duve C. The large-scale separation of peroxisomes, mitochondria, and lysosomes from the livers of rats injected with triton WR-1339. Improved isolation procedures, automated analysis, biochemical and morphological properties of fractions. J. Cell Biol., 37, 482–513 (1968).
- 5) Baudhuin P. Liver peroxisomes, cytology and function. Ann. N. Y. Acad. Sci., 168 (2 The Nature an), 214–228 (1969).
- 6) Reddy JK, Krishnakantha TP. Hepatic peroxisome proliferation: induction by two novel compounds structurally unrelated to clofibrate. Science, 190, 787–789 (1975).
- 7) Lazarow PB, De Duve C. A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. U.S.A., 73, 2043–2046 (1976).
- 8) Osumi T, Hashimoto T. Enhancement of fatty acyl-CoA oxidizing activity in rat liver peroxisomes by di-(i-ethylhexyl)phthalate. J. Biochem., 83, 1361–1365 (1978).
- 9) Bowen P, Lee CS, Zellweger H, Lindenberg R. A familial syndrome of multiple congenital defects. Bull. Johns Hopkins Hosp., 114, 402–414 (1964).
- 10) Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsamis MP, Wisniewski HK, Ritch RH, Norton WT, Rapin I, Gartner LM. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science, 182, 62–64 (1973).
- 11) Berger J, Dorninger F, Forss-Petter S, Kunze M. Peroxisomes in brain development and function. Biochim. Biophys. Acta, 1863, 934–955 (2016).
- 12) Waterham HR, Ferdinandusse S, Wanders RJ. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta, 1863, 922–933 (2016).
- 13) Vamecq J, Cherkaoui-Malki M, Andreoletti P, Latruffe N. The human peroxisome in health and disease: the story of an oddity becoming a vital organelle. Biochimie, 98, 4–15 (2014).
- 14) Haanstra JR, Gonzalez-Marcano EB, Gualdron-Lopez M, Michels PA. Biogenesis, maintenance and dynamics of glycosomes in trypanosomatid parasites. Biochim. Biophys. Acta, 1863, 1038–1048 (2016).
- 15) Graham IA. Seed storage oil mobilization. Annu. Rev. Plant Biol., 59, 115–142 (2008).
- 16) Steinberg G, Harmer NJ, Schuster M, Kilaru S. Woronin body-based sealing of septal pores. Fungal Genet. Biol., 109, 53–55 (2017).
- 17) Novikoff PM, Novikoff AB. Peroxisomes in absorptive cells of mammalian small intestine. J. Cell Biol., 53, 532–560 (1972).
- 18) Higashi T, Peters T Jr. Studies on rat liver catalase. I. combined immunochemical and enzymatic determination of catalase in liver cell fractions. J. Biol. Chem., 238, 3945–3951 (1963).
- 19) Imanaka T, Small GM, Lazarow PB. Translocation of acyl-CoA oxidase into peroxisomes requires ATP hydrolysis but not a membrane potential. J. Cell Biol., 105, 2915–2922 (1987).
- 20) Lazarow PB, Fujiki Y. Biogenesis of peroxisomes. Annu. Rev. Cell Biol., 1, 489–530 (1985).
- 21) Agrawal G, Subramani S. De novo peroxisome biogenesis: Evolving concepts and conundrums. Biochim. Biophys. Acta, 1863, 892–901 (2016).
- 22) Sugiura A, Mattie S, Prudent J, McBride HM. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature, 542, 251–254 (2017).
- 23) Moser AE, Singh I, Brown FR 3rd, Solish GI, Kelley RI, Benke PJ, Moser HW. The cerebrohepatorenal (Zellweger) syndrome. Increased levels and impaired degradation of very-long-chain fatty acids and their use in prenatal diagnosis. N. Engl. J. Med., 310, 1141–1146 (1984).
- 24) Datta NS, Wilson GN, Hajra AK. Deficiency of enzymes catalyzing the biosynthesis of glycerol-ether lipids in Zellweger syndrome. A new category of metabolic disease involving the absence of peroxisomes. N. Engl. J. Med., 311, 1080–1083 (1984).
- 25) Kase BF, Bjorkhem I, Haga P, Pedersen JI. Defective peroxisomal cleavage of the C27-steroid side chain in the cerebro-hepato-renal syndrome of Zellweger. J. Clin. Invest., 75, 427–435 (1985).
- 26) Gould SG, Keller GA, Subramani S. Identification of a peroxisomal targeting signal at the carboxy terminus of firefly luciferase. J. Cell Biol., 105, 2923–2931 (1987).
- 27) Gould SJ, Keller GA, Subramani S. Identification of peroxisomal targeting signals located at the carboxy terminus of four peroxisomal proteins. J. Cell Biol., 107, 897–905 (1988).
- 28) Osumi T, Tsukamoto T, Hata S, Yokota S, Miura S, Fujiki Y, Hijikata M, Miyazawa S, Hashimoto T. Amino-terminal presequence of the precursor of peroxisomal 3-ketoacyl-CoA thiolase is a cleavable signal peptide for peroxisomal targeting. Biochem. Biophys. Res. Commun., 181, 947–954 (1991).
- 29) Swinkels BW, Gould SJ, Bodnar AG, Rachubinski RA, Subramani S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J., 10, 3255–3262 (1991).
- 30) Rucktäschel R, Girzalsky W, Erdmann R. Protein import machineries of peroxisomes. Biochim. Biophys. Acta, 1808, 892–900 (2011).
- 31) Hasan S, Platta HW, Erdmann R. Import of proteins into the peroxisomal matrix. Front. Physiol., 4, 261 (2013).
- 32) Francisco T, Rodrigues TA, Pinto MP, Carvalho AF, Azevedo JE, Grou CP. Ubiquitin in the peroxisomal protein import pathway. Biochimie, 98, 29–35 (2014).
- 33) Fujiki Y, Nashiro C, Miyata N, Tamura S, Okumoto K. New insights into dynamic and functional assembly of the AAA peroxins, Pex1p and Pex6p, and their membrane receptor Pex26p in shuttling of PTS1-receptor Pex5p during peroxisome biogenesis. Biochim. Biophys. Acta, 1823, 145–149 (2012).
- 34) Francisco T, Rodrigues TA, Dias AF, Barros-Barbosa A, Bicho D, Azevedo JE. Protein transport into peroxisomes: Knowns and unknowns. BioEssays, 39, 1700047 (2017).
- 35) Santos MJ, Imanaka T, Shio H, Lazarow PB. Peroxisomal integral membrane proteins in control and Zellweger fibroblasts. J. Biol. Chem., 263, 10502–10509 (1988).
- 36) Santos MJ, Imanaka T, Shio H, Small GM, Lazarow PB. Peroxisomal membrane ghosts in Zellweger syndrome–aberrant organelle assembly. Science, 239, 1536–1538 (1988).
- 37) Imanaka T, Shiina Y, Takano T, Hashimoto T, Osumi T. Insertion of the 70-kDa peroxisomal membrane protein into peroxisomal membranes in vivo and in vitro. J. Biol. Chem., 271, 3706–3713 (1996).
- 38) Fang Y, Morrell JC, Jones JM, Gould SJ. PEX3 functions as a PEX19 docking factor in the import of class I peroxisomal membrane proteins. J. Cell Biol., 164, 863–875 (2004).
- 39) Shibata H, Kashiwayama Y, Imanaka T, Kato H. Domain architecture and activity of human Pex19p, a chaperone-like protein for intracellular trafficking of peroxisomal membrane proteins. J. Biol. Chem., 279, 38486–38494 (2004).
- 40) Sato Y, Shibata H, Nakatsu T, Nakano H, Kashiwayama Y, Imanaka T, Kato H. Structural basis for docking of peroxisomal membrane protein carrier Pex19p onto its receptor Pex3p. EMBO J., 29, 4083–4093 (2010).
- 41) Sato Y, Shibata H, Nakano H, Matsuzono Y, Kashiwayama Y, Kobayashi Y, Fujiki Y, Imanaka T, Kato H. Characterization of the interaction between recombinant human peroxin Pex3p and Pex19p: identification of TRP-104 IN Pex3p as a critical residue for the interaction. J. Biol. Chem., 283, 6136–6144 (2008).
- 42) Emmanouilidis L, Schutz U, Tripsianes K, Madl T, Radke J, Rucktaschel R, Wilmanns M, Schliebs W, Erdmann R, Sattler M. Allosteric modulation of peroxisomal membrane protein recognition by farnesylation of the peroxisomal import receptor PEX19. Nat. Commun., 8, 14635 (2017).
- 43) Fujiki Y, Okumoto K, Mukai S, Honsho M, Tamura S. Peroxisome biogenesis in mammalian cells. Front. Physiol., 5, 307 (2014).
- 44) Kikuchi M, Hatano N, Yokota S, Shimozawa N, Imanaka T, Taniguchi H. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem., 279, 421–428 (2004).
- 45) Morita M, Kurochkin IV, Motojima K, Goto S, Takano T, Okamura S, Sato R, Yokota S, Imanaka T. Insulin-degrading enzyme exists inside of rat liver peroxisomes and degrades oxidized proteins. Cell Struct. Funct., 25, 309–315 (2000).
- 46) Nakagawa T, Imanaka T, Morita M, Ishiguro K, Yurimoto H, Yamashita A, Kato N, Sakai Y. Peroxisomal membrane protein Pmp47 is essential in the metabolism of middle-chain fatty acid in yeast peroxisomes and Is associated with peroxisome proliferation. J. Biol. Chem., 275, 3455–3461 (2000).
- 47) Wanders RJ. Metabolic functions of peroxisomes in health and disease. Biochimie, 98, 36–44 (2014).
- 48) Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev Biol., 3, 83 (2015).
- 49) Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes are signaling platforms for antiviral innate immunity. Cell, 141, 668–681 (2010).
- 50) Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, Yang H, Miao HH, Li BL, Song BL. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell, 161, 291–306 (2015).
- 51) Dassa E, Bouige P. The ABC of ABCS: a phylogenetic and functional classification of ABC systems in living organisms. Res. Microbiol., 152, 211–229 (2001).
- 52) Vasiliou V, Vasiliou K, Nebert DW. Human ATP-binding cassette (ABC) transporter family. Hum. Genomics, 3, 281–290 (2009).
- 53) Kamijo K, Taketani S, Yokota S, Osumi T, Hashimoto T. The 70-kDa peroxisomal membrane protein is a member of the Mdr (P-glycoprotein)-related ATP-binding protein superfamily. J. Biol. Chem., 265, 4534–4540 (1990).
- 54) Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H, Poustka AM, Mandel JL, Aubourg P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature, 361, 726–730 (1993).
- 55) Lombard-Platet G, Savary S, Sarde CO, Mandel JL, Chimini G. A close relative of the adrenoleukodystrophy (ALD) gene codes for a peroxisomal protein with a specific expression pattern. Proc. Natl. Acad. Sci. U.S.A., 93, 1265–1269 (1996).
- 56) Holzinger A, Kammerer S, Berger J, Roscher AA. cDNA cloning and mRNA expression of the human adrenoleukodystrophy related protein (ALDRP), a peroxisomal ABC transporter. Biochem. Biophys. Res. Commun., 239, 261–264 (1997).
- 57) Holzinger A, Kammerer S, Roscher AA. Primary structure of human PMP69, a putative peroxisomal ABC-transporter. Biochem. Biophys. Res. Commun., 237, 152–157 (1997).
- 58) Shani N, Jimenez-Sanchez G, Steel G, Dean M, Valle D. Identification of a fourth half ABC transporter in the human peroxisomal membrane. Hum. Mol. Genet., 6, 1925–1931 (1997).
- 59) Higgins CF. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol., 8, 67–113 (1992).
- 60) Contreras M, Sengupta TK, Sheikh F, Aubourg P, Singh I. Topology of ATP-binding domain of adrenoleukodystrophy gene product in peroxisomes. Arch. Biochem. Biophys., 334, 369–379 (1996).
- 61) Kashiwayama Y, Morita M, Kamijo K, Imanaka T. Nucleotide-induced conformational changes of PMP70, an ATP binding cassette transporter on rat liver peroxisomal membranes. Biochem. Biophys. Res. Commun., 291, 1245–1251 (2002).
- 62) Tanaka AR, Tanabe K, Morita M, Kurisu M, Kasiwayama Y, Matsuo M, Kioka N, Amachi T, Imanaka T, Ueda K. ATP binding/hydrolysis by and phosphorylation of peroxisomal ATP-binding cassette proteins PMP70 (ABCD3) and adrenoleukodystrophy protein (ABCD1). J. Biol. Chem., 277, 40142–40147 (2002).
- 63) Guimarães CP, Sá-Miranda C, Azevedo JE. Probing substrate-induced conformational alterations in adrenoleukodystrophy protein by proteolysis. J. Hum. Genet., 50, 99–105 (2005).
- 64) Okamoto T, Kawaguchi K, Watanabe S, Agustina R, Ikejima T, Ikeda K, Nakano M, Morita M, Imanaka T. Characterization of human ATP-binding cassette protein subfamily D reconstituted into proteoliposomes. Biochem. Biophys. Res. Commun., 496, 1122–1127 (2018).
- 65) Liu LX, Janvier K, Berteaux-Lecellier V, Cartier N, Benarous R, Aubourg P. Homo- and heterodimerization of peroxisomal ATP-binding cassette half-transporters. J. Biol. Chem., 274, 32738–32743 (1999).
- 66) Guimarães CP, Domingues P, Aubourg P, Fouquet F, Pujol A, Jimenez-Sanchez G, Sa-Miranda C, Azevedo JE. Mouse liver PMP70 and ALDP: homomeric interactions prevail in vivo. Biochim. Biophys. Acta, 1689, 235–243 (2004).
- 67) Hillebrand M, Verrier SE, Ohlenbusch A, Schafer A, Soling HD, Wouters FS, Gartner J. Live cell FRET microscopy: homo- and heterodimerization of two human peroxisomal ABC transporters, the adrenoleukodystrophy protein (ALDP, ABCD1) and PMP70 (ABCD3). J. Biol. Chem., 282, 26997–27005 (2007).
- 68) Geillon F, Gondcaille C, Raas Q, Dias AMM, Pecqueur D, Truntzer C, Lucchi G, Ducoroy P, Falson P, Savary S, Trompier D. Peroxisomal ATP-binding cassette transporters form mainly tetramers. J. Biol. Chem., 292, 6965–6977 (2017).
- 69) Lee A, Asahina K, Okamoto T, Kawaguchi K, Kostsin DG, Kashiwayama Y, Takanashi K, Yazaki K, Imanaka T, Morita M. Role of NH2-terminal hydrophobic motif in the subcellular localization of ATP-binding cassette protein subfamily D: common features in eukaryotic organisms. Biochem. Biophys. Res. Commun., 453, 612–618 (2014).
- 70) Morita M, Imanaka T. Peroxisomal ABC transporters: structure, function and role in disease. Biochim. Biophys. Acta, 1822, 1387–1396 (2012).
- 71) Kashiwayama Y, Seki M, Yasui A, Murasaki Y, Morita M, Yamashita Y, Sakaguchi M, Tanaka Y, Imanaka T. 70-kDa peroxisomal membrane protein related protein (P70R/ABCD4) localizes to endoplasmic reticulum not peroxisomes, and NH2-terminal hydrophobic property determines the subcellular localization of ABC subfamily D proteins. Exp. Cell Res., 315, 190–205 (2009).
- 72) Kawaguchi K, Okamoto T, Morita M, Imanaka T. Translocation of the ABC transporter ABCD4 from the endoplasmic reticulum to lysosomes requires the escort protein LMBD1. Sci. Rep., 6, 30183 (2016).
- 73) Sacksteder KA, Jones JM, South ST, Li X, Liu Y, Gould SJ. PEX19 binds multiple peroxisomal membrane proteins, is predominantly cytoplasmic, and is required for peroxisome membrane synthesis. J. Cell Biol., 148, 931–944 (2000).
- 74) Biermanns M, Gartner J. Targeting elements in the amino-terminal part direct the human 70-kDa peroxisomal integral membrane protein (PMP70) to peroxisomes. Biochem. Biophys. Res. Commun., 285, 649–655 (2001).
- 75) Kashiwayama Y, Asahina K, Shibata H, Morita M, Muntau AC, Roscher AA, Wanders RJ, Shimozawa N, Sakaguchi M, Kato H, Imanaka T. Role of Pex19p in the targeting of PMP70 to peroxisome. Biochim. Biophys. Acta, 1746, 116–128 (2005).
- 76) Kashiwayama Y, Asahina K, Morita M, Imanaka T. Hydrophobic regions adjacent to transmembrane domains 1 and 5 are important for the targeting of the 70-kDa peroxisomal membrane protein. J. Biol. Chem., 282, 33831–33844 (2007).
- 77) Rottensteiner H, Kramer A, Lorenzen S, Stein K, Landgraf C, Volkmer-Engert R, Erdmann R. Peroxisomal membrane proteins contain common Pex19p-binding sites that are an integral part of their targeting signals. Mol. Biol. Cell, 15, 3406–3417 (2004).
- 78) Halbach A, Lorenzen S, Landgraf C, Volkmer-Engert R, Erdmann R, Rottensteiner H. Function of the PEX19-binding site of human adrenoleukodystrophy protein as targeting motif in man and yeast. PMP targeting is evolutionarily conserved. J. Biol. Chem., 280, 21176–21182 (2005).
- 79) Tseng LT, Lin CL, Tzen KY, Chang SC, Chang MF. LMBD1 protein serves as a specific adaptor for insulin receptor internalization. J. Biol. Chem., 288, 32424–32432 (2013).
- 80) Iwashita S, Tsuchida M, Tsukuda M, Yamashita Y, Emi Y, Kida Y, Komori M, Kashiwayama Y, Imanaka T, Sakaguchi M. Multiple organelle-targeting signals in the N-terminal portion of peroxisomal membrane protein PMP70. J. Biochem., 147, 581–590 (2010).
- 81) van Roermund CW, Visser WF, Ijlst L, Waterham HR, Wanders RJ. Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid β-oxidation. Biochim. Biophys. Acta, 1811, 148–152 (2011).
- 82) van Roermund CW, Ijlst L, Wagemans T, Wanders RJ, Waterham HR. A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim. Biophys. Acta, 1841, 563–568 (2014).
- 83) Ferdinandusse S, Jimenez-Sanchez G, Koster J, Denis S, Van Roermund CW, Silva-Zolezzi I, Moser AB, Visser WF, Gulluoglu M, Durmaz O, Demirkol M, Waterham HR, Gokcay G, Wanders RJ, Valle D. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet., 24, 361–370 (2015).
- 84) Coelho D, Kim JC, Miousse IR, Fung S, du Moulin M, Buers I, Suormala T, Burda P, Frapolli M, Stucki M, Nurnberg P, Thiele H, Robenek H, Hohne W, Longo N, Pasquali M, Mengel E, Watkins D, Shoubridge EA, Majewski J, Rosenblatt DS, Fowler B, Rutsch F, Baumgartner MR. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet., 44, 1152–1155 (2012).
- 85) Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun., 3, 925 (2012).
- 86) Cartier N, Lopez J, Moullier P, Rocchiccioli F, Rolland MO, Jorge P, Mosser J, Mandel JL, Bougneres PF, Danos O, Aubourg P. Retroviral-mediated gene transfer corrects very-long-chain fatty acid metabolism in adrenoleukodystrophy fibroblasts. Proc. Natl. Acad. Sci. U.S.A., 92, 1674–1678 (1995).
- 87) Braiterman LT, Zheng S, Watkins PA, Geraghty MT, Johnson G, McGuinness MC, Moser AB, Smith KD. Suppression of peroxisomal membrane protein defects by peroxisomal ATP binding cassette (ABC) proteins. Hum. Mol. Genet., 7, 239–247 (1998).
- 88) De Marcos Lousa C, van Roermund CW, Postis VL, Dietrich D, Kerr ID, Wanders RJ, Baldwin SA, Baker A, Theodoulou FL. Intrinsic acyl-CoA thioesterase activity of a peroxisomal ATP binding cassette transporter is required for transport and metabolism of fatty acids. Proc. Natl. Acad. Sci. U.S.A., 110, 1279–1284 (2013).
- 89) van Roermund CW, Ijlst L, Majczak W, Waterham HR, Folkerts H, Wanders RJ, Hellingwerf KJ. Peroxisomal fatty acid uptake mechanism in Saccharomyces cerevisiae. J. Biol. Chem., 287, 20144–20153 (2012).
- 90) Wiesinger C, Kunze M, Regelsberger G, Forss-Petter S, Berger J. Impaired very long-chain acyl-CoA β-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J. Biol. Chem., 288, 19269–19279 (2013).
- 91) Alexson SE, Mentlein R, Wernstedt C, Hellman U. Isolation and characterization of microsomal acyl-CoA thioesterase. A member of the rat liver microsomal carboxylesterase multi-gene family. Eur. J. Biochem., 214, 719–727 (1993).
- 92) Svensson LT, Alexson SE, Hiltunen JK. Very long chain and long chain acyl-CoA thioesterases in rat liver mitochondria. Identification, purification, characterization, and induction by peroxisome proliferators. J. Biol. Chem., 270, 12177–12183 (1995).
- 93) Yamada J, Kurata A, Hirata M, Taniguchi T, Takama H, Furihata T, Shiratori K, Iida N, Takagi-Sakuma M, Watanabe T, Kurosaki K, Endo T, Suga T. Purification, molecular cloning, and genomic organization of human brain long-chain acyl-CoA hydrolase. J. Biochem., 126, 1013–1019 (1999).
- 94) Huhtinen K, O’Byrne J, Lindquist PJ, Contreras JA, Alexson SE. The peroxisome proliferator-induced cytosolic type I acyl-CoA thioesterase (CTE-I) is a serine-histidine-aspartic acid α/β hydrolase. J. Biol. Chem., 277, 3424–3432 (2002).
- 95) Hunt MC, Tillander V, Alexson SE. Regulation of peroxisomal lipid metabolism: the role of acyl-CoA and coenzyme A metabolizing enzymes. Biochimie, 98, 45–55 (2014).
- 96) Pujol A, Ferrer I, Camps C, Metzger E, Hindelang C, Callizot N, Ruiz M, Pampols T, Giros M, Mandel JL. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum. Mol. Genet., 13, 2997–3006 (2004).
- 97) Fourcade S, Ruiz M, Camps C, Schluter A, Houten SM, Mooyer PA, Pampols T, Dacremont G, Wanders RJ, Giros M, Pujol A. A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am. J. Physiol. Endocrinol. Metab., 296, E211–E221 (2009).
- 98) Genin EC, Geillon F, Gondcaille C, Athias A, Gambert P, Trompier D, Savary S. Substrate specificity overlap and interaction between adrenoleukodystrophy protein (ALDP/ABCD1) and adrenoleukodystrophy-related protein (ALDRP/ABCD2). J. Biol. Chem., 286, 8075–8084 (2011).
- 99) Fourcade S, Ruiz M, Guilera C, Hahnen E, Brichta L, Naudi A, Portero-Otin M, Dacremont G, Cartier N, Wanders R, Kemp S, Mandel JL, Wirth B, Pamplona R, Aubourg P, Pujol A. Valproic acid induces antioxidant effects in X-linked adrenoleukodystrophy. Hum. Mol. Genet., 19, 2005–2014 (2010).
- 100) Imanaka T, Aihara K, Takano T, Yamashita A, Sato R, Suzuki Y, Yokota S, Osumi T. Characterization of the 70-kDa peroxisomal membrane protein, an ATP binding cassette transporter. J. Biol. Chem., 274, 11968–11976 (1999).
- 101) Morita M, Shinbo S, Asahi A, Imanaka T. Very long chain fatty acid β-oxidation in astrocytes: contribution of the ABCD1-dependent and -independent pathways. Biol. Pharm. Bull., 35, 1972–1979 (2012).
- 102) Coelho D, Suormala T, Stucki M, Lerner-Ellis JP, Rosenblatt DS, Newbold RF, Baumgartner MR, Fowler B. Gene identification for the cblD defect of vitamin B12 metabolism. N. Engl. J. Med., 358, 1454–1464 (2008).
- 103) Rutsch F, Gailus S, Miousse IR, Suormala T, Sagne C, Toliat MR, Nurnberg G, Wittkampf T, Buers I, Sharifi A, Stucki M, Becker C, Baumgartner M, Robenek H, Marquardt T, Hohne W, Gasnier B, Rosenblatt DS, Fowler B, Nurnberg P. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet., 41, 234–239 (2009).
- 104) Beedholm-Ebsen R, van de Wetering K, Hardlei T, Nexo E, Borst P, Moestrup SK. Identification of multidrug resistance protein 1 (MRP1/ABCC1) as a molecular gate for cellular export of cobalamin. Blood, 115, 1632–1639 (2010).
- 105) Borths EL, Poolman B, Hvorup RN, Locher KP, Rees DC. In vitro functional characterization of BtuCD-F, the Escherichia coli ABC transporter for vitamin B12 uptake. Biochemistry, 44, 16301–16309 (2005).
- 106) Wanders RJ, Klouwer FC, Ferdinandusse S, Waterham HR, Poll-The BT. Clinical and Laboratory Diagnosis of Peroxisomal Disorders. Methods Mol. Biol., 1595, 329–342 (2017).
- 107) Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med., 356, 1736–1741 (2007).
- 108) Ebberink MS, Koster J, Visser G, Spronsen F, Stolte-Dijkstra I, Smit GP, Fock JM, Kemp S, Wanders RJ, Waterham HR. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11β gene. J. Med. Genet., 49, 307–313 (2012).
- 109) Huber N, Guimaraes S, Schrader M, Suter U, Niemann A. Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission. EMBO Rep., 14, 545–552 (2013).
- 110) Wang W, Xia ZJ, Farre JC, Subramani S. TRIM37, a novel E3 ligase for PEX5-mediated peroxisomal matrix protein import. J. Cell Biol., 216, 2843–2858 (2017).
- 111) Ferdinandusse S, Falkenberg KD, Koster J, Mooyer PA, Jones R, van Roermund CWT, Pizzino A, Schrader M, Wanders RJA, Vanderver A, Waterham HR. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J. Med. Genet., 54, 330–337 (2017).
- 112) Yagita Y, Shinohara K, Abe Y, Nakagawa K, Al-Owain M, Alkuraya FS, Fujiki Y. Deficiency of a Retinal Dystrophy Protein, Acyl-CoA Binding Domain-containing 5 (ACBD5), Impairs Peroxisomal β-Oxidation of Very-long-chain Fatty Acids. J. Biol. Chem., 292, 691–705 (2017).
- 113) Hua R, Cheng D, Coyaud E, Freeman S, Di Pietro E, Wang Y, Vissa A, Yip CM, Fairn GD, Braverman N, Brumell JH, Trimble WS, Raught B, Kim PK. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol., 216, 367–377 (2017).
- 114) Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD, Kass NE, Moser HW. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann. Neurol., 49, 512–517 (2001).
- 115) Takemoto Y, Suzuki Y, Tamakoshi A, Onodera O, Tsuji S, Hashimoto T, Shimozawa N, Orii T, Kondo N. Epidemiology of X-linked adrenoleukodystrophy in Japan. J. Hum. Genet., 47, 590–593 (2002).
- 116) Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, Poll-The BT. X-Linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis., 7, 51 (2012).
- 117) Trompier D, Savary S. X-Linked adrenoleukodystrophy. Colloquium series on the genetic basis of human disease. Morgan & Claypool Life Sciences, San Rafael, CA, U.S.A. (2013).
- 118) Siemerling E, Creutzfeldt HG. Bronzekrankheit und sklerosierende Encephalomyelitis diffuse sklerose. Eur. Arch. Psychiatry Clin. Neurosci., 68, 217–244 (1923).
- 119) Fanconi A, Prader A, Isler W, Luethy F, Siebenmann R. Addison’s disease with cerebral sclerosis in childhood. a hereditary syndrome transmitted through chromosome X? Helv. Paediatr. Acta, 18, 480–501 (1963).
- 120) Blaw ME. Melanodermic type leukodystrophy (adrenoleukodystrophy). Neurodystrophies and neurolipidoses. (Vinken PJ, Bruyn GW, eds.) North Holland Publishing Company, Amsterdam, pp. 128–133 (1970).
- 121) Igarashi M, Schaumburg HH, Powers J, Kishmoto Y, Kolodny E, Suzuki K. Fatty acid abnormality in adrenoleukodystrophy. J. Neurochem., 26, 851–860 (1976).
- 122) van de Beek MC, Dijkstra IM, van Lenthe H, Ofman R, Goldhaber-Pasillas D, Schauer N, Schackmann M, Engelen-Lee JY, Vaz FM, Kulik W, Wanders RJ, Engelen M, Kemp S. C26:0-Carnitine Is a New Biomarker for X-Linked Adrenoleukodystrophy in Mice and Man. PLoS ONE, 11, e0154597 (2016).
- 123) Kemp S, Berger J, Aubourg P. X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim. Biophys. Acta, 1822, 1465–1474 (2012).
- 124) Berger J, Forss-Petter S, Eichler FS. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie, 98, 135–142 (2014).
- 125) Fourcade S, Lopez-Erauskin J, Ruiz M, Ferrer I, Pujol A. Mitochondrial dysfunction and oxidative damage cooperatively fuel axonal degeneration in X-linked adrenoleukodystrophy. Biochimie, 98, 143–149 (2014).
- 126) Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl. Clin. Genet., 8, 109–121 (2015).
- 127) Peters C, Charnas LR, Tan Y, Ziegler RS, Shapiro EG, DeFor T, Grewal SS, Orchard PJ, Abel SL, Goldman AI, Ramsay NK, Dusenbery KE, Loes DJ, Lockman LA, Kato S, Aubourg PR, Moser HW, Krivit W. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood, 104, 881–888 (2004).
- 128) Morita M, Honda A, Kobayashi A, Watanabe Y, Watanabe S, Kawaguchi K, Takashima S, Shimozawa N, Imanaka T. Effect of Lorenzo’s Oil on Hepatic Gene Expression and the Serum Fatty Acid Level in abcd1-Deficient Mice. JIMD Rep., 38, 67–74 (2018).
- 129) Morita M, Shimozawa N, Kashiwayama Y, Suzuki Y, Imanaka T. ABC subfamily D proteins and very long chain fatty acid metabolism as novel targets in adrenoleukodystrophy. Curr. Drug Targets, 12, 694–706 (2011).
- 130) Zhang X, De Marcos Lousa C, Schutte-Lensink N, Ofman R, Wanders RJ, Baldwin SA, Baker A, Kemp S, Theodoulou FL. Conservation of targeting but divergence in function and quality control of peroxisomal ABC transporters: an analysis using cross-kingdom expression. Biochem. J., 436, 547–557 (2011).
- 131) Morita M, Matsumoto S, Sato A, Inoue K, Kostsin DG, Yamazaki K, Kawaguchi K, Shimozawa N, Kemp S, Wanders RJ, Kojima H, Okabe T, Imanaka T. Stability of the ABCD1 protein with a missense mutation: a novel approach to finding therapeutic compounds for X-linked adrenoleukodystrophy. JIMD Rep., 44, 23–31 (2019).
- 132) Morita M, Kanai M, Mizuno S, Iwashima M, Hayashi T, Shimozawa N, Suzuki Y, Imanaka T. Baicalein 5,6,7-trimethyl ether activates peroxisomal but not mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis., 31, 442–449 (2008).
- 133) Morita M, Matsumoto S, Okazaki A, Tomita K, Watanabe S, Kawaguchi K, Minato D, Matsuya Y, Shimozawa N, Imanaka T. A novel method for determining peroxisomal fatty acid β-oxidation. J. Inherit. Metab. Dis., 39, 725–731 (2016).
- 134) Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrere F, Blanche S, Audit M, Payen E, Leboulch P, l’Homme B, Bougneres P, Von Kalle C, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science, 326, 818–823 (2009).
- 135) Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, Armant M, Dansereau C, Lund TC, Miller WP, Raymond GV, Sankar R, Shah AJ, Sevin C, Gaspar HB, Gissen P, Amartino H, Bratkovic D, Smith NJC, Paker AM, Shamir E, O’Meara T, Davidson D, Aubourg P, Williams DA. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med., 377, 1630–1638 (2017).
- 136) Hama K, Nagai T, Nishizawa C, Ikeda K, Morita M, Satoh N, Nakanishi H, Imanaka T, Shimozawa N, Taguchi R, Inoue K, Yokoyama K. Molecular species of phospholipids with very long chain fatty acids in skin fibroblasts of Zellweger syndrome. Lipids, 48, 1253–1267 (2013).
- 137) Hama K, Fujiwara Y, Morita M, Yamazaki F, Nakashima Y, Takei S, Takashima S, Setou M, Shimozawa N, Imanaka T, Yokoyama K. Profiling and Imaging of Phospholipids in Brains of Abcd1-Deficient Mice. Lipids, 53, 85–102 (2018).
- 138) Herzog K, Pras-Raves ML, Ferdinandusse S, Vervaart MAT, Luyf ACM, van Kampen AHC, Wanders RJA, Waterham HR, Vaz FM. Functional characterisation of peroxisomal β-oxidation disorders in fibroblasts using lipidomics. J. Inherit. Metab. Dis., 41, 479–487 (2018).
- 139) Jang J, Kang HC, Kim HS, Kim JY, Huh YJ, Kim DS, Yoo JE, Lee JA, Lim B, Lee J, Yoon TM, Park IH, Hwang DY, Daley GQ, Kim DW. Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann. Neurol., 70, 402–409 (2011).
- 140) Jang J, Park S, Jin Hur H, Cho HJ, Hwang I, Pyo Kang Y, Im I, Lee H, Lee E, Yang W, Kang HC, Won Kwon S, Yu JW, Kim DW. 25-Hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat. Commun., 7, 13129 (2016).
- 141) You YR, Son D, Kang PJ, You S, Kim DS. Generation of induced pluripotent stem cell (iPSC) line from a 21-year-old X-linked adrenoleukodystrophy (X-ALD) patient. Stem Cell Res., 25, 136–138 (2017).
- 142) Lieber DS, Hershman SG, Slate NG, Calvo SE, Sims KB, Schmahmann JD, Mootha VK. Next generation sequencing with copy number variant detection expands the phenotypic spectrum of HSD17B4-deficiency. BMC Med. Genet., 15, 30 (2014).
- 143) Renaud M, Guissart C, Mallaret M, Ferdinandusse S, Cheillan D, Drouot N, Muller J, Claustres M, Tranchant C, Anheim M, Koenig M. Expanding the spectrum of PEX10-related peroxisomal biogenesis disorders: slowly progressive recessive ataxia. J. Neurol., 263, 1552–1558 (2016).