Regular Articles
Protein Tyrosine Phosphatase Inhibitor, Orthovanadate, Induces Contraction via Rho Kinase Activation in Mouse Thoracic Aortas
2019 年 42 巻 6 号 p. 877-885
詳細
2019 年 42 巻 6 号 p. 877-885
Orthovanadate (OVA), a protein tyrosine phosphatase inhibitor, induces contraction in endothelium-denuded mouse thoracic aortas. OVA-induced contraction was significantly (vs. control rings) suppressed by Rho kinase (Y-27632, 10 µM), extracellular signal-regulated kinase 1 and 2 (Erk1/2, FR180204, 10 µM), Erk1/2 kinase (MEK, PD98059, 10 µM), epidermal growth factor receptor (EGFR, AG1478, 10 µM), and Src inhibitors, and was partially suppressed by c-Jun N-terminal kinase (JNK, AS601245, 10 µM) and p38 (SB203580, 10 µM) inhibitors. However, a myosin light chain kinase inhibitor (ML-7, 10 µM) and a metalloproteinase inhibitor (TAPI-0, 10 µM) had no effect on OVA-induced contraction in mouse thoracic aortas. Phosphorylation of myosin phosphatase target subunit 1 (MYPT1) was abolished by inhibitors of Src, EGFR, MEK, Erk1/2, and Rho kinase, but not by inhibitors of JNK and p38. Erk1/2 phosphorylation by OVA was blocked by inhibitors of EGFR, Src, MEK, and Erk1/2, but not by Rho kinase inhibition. Src phosphorylation at Tyr-416 was abrogated by only Src inhibitor. EGFR phosphorylation at Tyr-1173 was suppressed by a Src inhibitor. These findings suggest that OVA induces contraction via activation of Src, EGFR, MEK, Erk1/2, and Rho kinase, leading to inactivation of myosin light chain phosphatase via MYPT1 phosphorylation.
Vanadium compounds have various biochemical and pharmacological properties because of the structural similarity of vanadium to phosphate. Several studies have proved that vanadates possess ATPase1) and protein tyrosine phosphatase (PTPase) inhibitory activities,2) epidermal growth factor (EGF)-like mitogenic activity,3) insulin-mimetic properties,4) antiapoptotic activities,5) and antitumor or carcinogenic properties.6,7) In addition, vanadates induce endothelium-dependent vasodilation in porcine coronary, renal, and femoral arteries, as well as in rat mesenteric arteries.8,9) These relaxations were abolished by pretreatment with the nitric oxide (NO) synthase (NOS) inhibitor, N(G)-nitro-L-arginine methyl ester (L-NAME). Moreover, sodium orthovanadate (OVA) induced NO release via Akt-induced endothelial NOS phosphorylation in bovine lung microvascular cells.10) In contrast, several studies reported that vanadium compounds, such as OVA and pervanadate, have vasocontractile effects11–16) and nonvascular smooth muscle contractile effects, such as those on guinea pig taenia coli, trachea, and gallbladder smooth muscle,17–19) and the rat gastric longitudinal muscle and myometrium.12,20) The contractile effects of vanadates are considered to be related to the inhibition of PTPases,12,17,18) but not ATPases,13,14) because genistein, a protein tyrosine kinase inhibitor, suppresses contraction by vanadates. In addition, the inhibition of Rho kinase with Y-27632 abrogated the contraction and phosphorylation of myosin light chain (MLC) by OVA in guinea pig ileal longitudinal smooth muscle.21) Our previous studies also showed that OVA exerts contractile response through phosphorylation of the myosin phosphatase target subunit 1 (MYPT1) of myosin light chain phosphatase (MLCP) by the activation of Rho kinase in the rat thoracic aortas and mesenteric arteries.22,23) These contractile effects were abolished by inhibition of Src, EGF receptor (EGFR), and Rho kinase in both arteries,22,23) and by inhibition of extracellular signal-regulated kinases 1 and 2 (Erk1/2) and Erk1/2 kinase (MEK) in rat mesenteric arteries.22) Furthermore, a metalloproteinase inhibitor, an inhibitor of heparin/EGF binding, and a Src inhibitor attenuated OVA-induced contraction and EGFR phosphorylation in the rat thoracic aortas.23) However, a metalloproteinase inhibitor and an inhibitor of heparin/EGF binding had no effect on OVA-induced contraction in mesenteric arteries.22) These findings indicate that OVA induces the activation of Src, which transactivates EGFR indirectly through the shedding of heparin-binding (HB)-EGF, and directly activates EGFR via Src-catalyzed phosphorylation of EGFR in the rat thoracic aortas, but only the direct pathway is utilized in mesenteric arteries. In these reports, it was considered that different artery sites participate in independent pathways of OVA-induced contraction. However, it is unknown whether OVA elicits contraction and phosphorylation of MYPT1 in mouse thoracic aortas. Therefore, we determine whether OVA induces contraction in mouse thoracic aortas. In addition, we investigated the possibility that this OVA-induced contraction is mediated by Src, EGFR, mitogen-activated protein kinases (MAPKs), and Rho kinase.
OVA was purchased from Nacalai Tesque (Kyoto, Japan). All inhibitors were purchased from Merck-Millipore (Tokyo, Japan). The inhibitors (product name, chemical name, target molecule, and used concentration) are described in Table 1. The concentration of inhibitors was selected based on our previous studies.22–25)
| Product name | Chemical name | Target molecule | Used concentration (µM) |
|---|---|---|---|
| AG1478 | 4-(3-Chloroanilino)-6,7-dimethoxyquinazoline | EGFR | 10 |
| AS601245 | (Z)-2-(Benzo[d]thiazol-2[3H]-ylidene)-2-(2-[{2-(pyridin-3-yl)ethyl}amino]pyrimidin-4-yl)acetonitrile | JNK | 10 |
| FR180204 | 5-(2-Phenyl-pyrazolo[1,5-a]pyridin-3-yl)-1H-pyrazolo[3,4-c]pyridazin-3-ylamine | Erk1/2 | 10 |
| ML-7 | 1-(5-Iodonaphthalene-1-sulfonyl)homopiperazine | MLCK | 10 |
| PD98059 | 2ʹ-Amino-3ʹ-methoxyflavone | MEK | 10 |
| PP2 | 4-Amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine | Src | 3 |
| SB203580 | 4-(4-Fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole | p38 MAPK | 10 |
| TAPI-0 | N-(R)-(2-[Hydroxyaminocarbonyl]methyl)-4-methylpentanoyl-L-naphthylalanyl-L-alanine amide | Metalloproteinase | 10 |
| Y-27632 | R-(+)-trans-N-(4-Pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide | Rho kinase | 10 |
All animal experiments were performed according to the guidelines of the Kobe Gakuin University Experimental Animal Care and Use Committee (16-03, 11 July 2016, A17-14, 6 April 2017). Male, 7-week-old ddY mice (Japan SLC, Hamamatsu, Japan) were used for all studies. Mice were euthanized by bleeding from the carotid arteries under ether anesthesia. The thoracic aortas were excised and immediately placed in Krebs–Henseleit solution of the following composition (mM): NaCl 118.4, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, glucose 11.1. The aortas were cleaned of adherent connective tissue and cut into rings (3 mm long). The endothelium was removed by carefully rotating a manipulator inside the lumen of the rings (confirmed by the absence of acetylcholine-induced relaxation under contraction elicited by phenylephrine). Each ring was fixed vertically under a resting tension of 0.5 g in a 5-mL organ chamber filled with the Krebs–Henseleit solution (37°C, pH 7.4) as described previously.26) The chamber solution was continuously aerated with a gas mixture of 95% O2/5% CO2, and the rings were allowed to equilibrate for 1 h before the start of the experiments. The isometric tension was measured as described previously.22–26) After equilibration, OVA was cumulatively added to the chamber solution to final concentration of between 31.3–500 µM. Inhibitors were dissolved in dimethyl sulfoxide and 10 µL of solution was added to the chamber 15 min prior to OVA. The contractile responses observed were expressed as a percentage of the maximal contraction evoked by 40 mM KCl. KCl was added before treatment of inhibitors. Typical traces obtained in organ chamber are presented in Fig. 1.
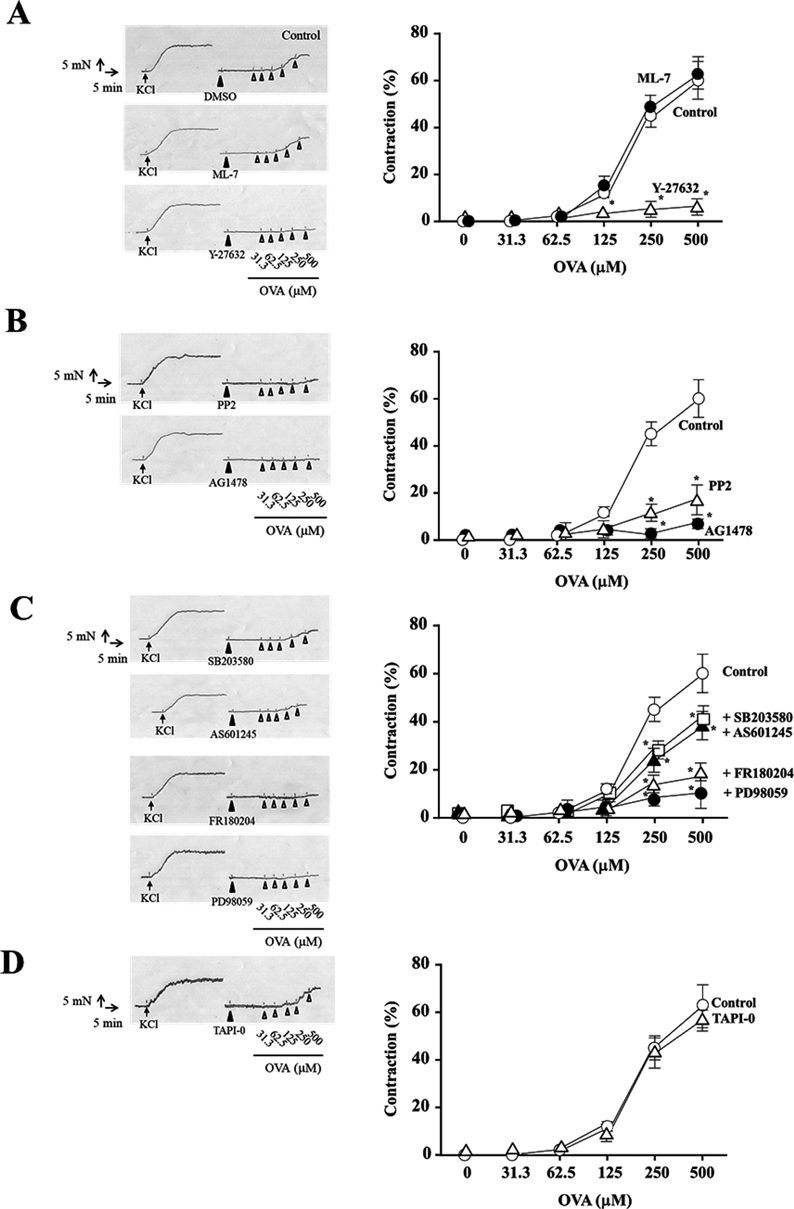
A: ML-7 (10 µM) or Y-27632 (10 µM); B: PP2 (3 µM) or AG1478 (10 µM); C: SB203580 (10 µM), AS601245 (10 µM), FR180204 (10 µM), or PD98059 (10 µM); D: TAPI-0 (10 µM) was added 15 min before OVA treatment. Cumulative contraction response curves normalized to 40 mM KCl-induced contraction. Corresponding traces are shown in the left of contraction response curves. Data are means ± S.E.M. for thoracic aortic rings from 4 mice, * p < 0.05 vs. control rings.
The endothelium-denuded aortic rings were equilibrated as described above and then treated with OVA (250 µM). In some experiments, inhibitors were added to the organ chamber 15 min before OVA treatment. After stimulation with OVA for 5 min, the rings were homogenized in 50 µL lysis buffer, comprised of 50 mM Tris–HCl (pH 7.4), 25 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), protease inhibitor cocktail (Nacalai Tesque), and phosphatase inhibitor cocktail (Nacalai Tesque). Samples were centrifuged at 15000 × g for 10 min at 4°C, and the concentration of soluble protein in the supernatant was measured by bicinchoninic acid (BCA) protein assay reagent (Thermo Scientific, Waltham, MA, U.S.A.). The 3 µg of protein was loaded per well and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to polyvinylidene fluoride (PVDF) membranes (Immobilon-P®; Millipore, Billerica, MA, U.S.A.). The blots were blocked with 5% skim milk in TTBS (10 mM Tris in 100 mM NaCl containing 0.1% Tween 20, pH 7.5). To specifically detect total or phosphorylated protein, blots were incubated with primary antibodies available commercially (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A. and Cell Signaling Technology, Danvers, MA, U.S.A.). The primary antibodies used in this study are included in Table 2. Then the bound antibodies were detected by peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) antibodies or anti-mouse IgG antibodies (β-actin) and the Chemi-Lumi One Super system (Chemi-Lumi One Super; Nacalai Tesque). Immunoblots were quantified using densitometry with VersaDoc™ 5000MP (Bio-Rad Laboratories, Hercules, CA, U.S.A.) and Quantity One software (Bio-Rad Laboratories).
| Antibody | Dilution | Catalog No. | Purchased from |
|---|---|---|---|
| MYPT1 | 1 : 200 | sc-25618 | Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.) |
| Thr-853-phosphorylated MYPT1 | 1 : 200 | sc-17432-R | Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.) |
| Erk1/2 | 1 : 1000 | #4695-P | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| Thr-202/Tyr-204-phosphorylated Erk1/2 | 1 : 1000 | #9101-S | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| p38 MAPK | 1 : 200 | sc-7149 | Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.) |
| Tyr-182-phosphorylated p38 MAPK | 1 : 200 | sc-7973 | Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.) |
| JNK | 1 : 1000 | #9252-S | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| Thr-183-phosphorylated JNK | 1 : 1000 | #9251-S | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| Src | 1 : 1000 | #2102-S | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| Tyr-416-phosphorylated Src | 1 : 1000 | #2101-S | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| EGFR | 1 : 500 | #2232 | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| Tyr-1173-phosphorylated EGFR | 1 : 500 | #4407 | Cell Signaling Technology (Danvers, MA, U.S.A.) |
| β-Actin | 1 : 5000 | A2228 | Bio-Rad Laboratories (Hercules, CA, U.S.A.) |
After stimulation by OVA, the plasma membrane proteins were extracted from the rings using the Plasma Membrane Protein Extraction Kit according to the manufacturer’s instructions (101 Bio., CA, U.S.A.). The phosphorylated EGFR and EGFR levels were measured by Western blotting as noted above. The blots were blocked with 1% bovine serum albumin in TTBS.
Statistical AnalysisAll data are expressed as mean ± standard error of the mean (S.E.M.). Statistical comparisons were performed using one-way ANOVA with pairwise comparisons performed using the Bonferroni–Dunn method. Concentration–response curves were compared using repeated-measures ANOVA followed by Bonferroni–Dunn test using the Graph Pad Prism 6 software. Differences were considered significant at p < 0.05.
We observed that OVA induced relaxation in endothelium-intact mouse thoracic aortic rings. The OVA-induced relaxation was blocked by the treatment with inhibitor of phosphoinositide 3-kinase and Src but not inhibitor of cyclooxygenase. Interestingly, treatment with NOS inhibitor (L-NAME) to endothelium-intact mouse thoracic aortic rings induced weak contraction (data not shown). Then, we focused on the mechanisms of OVA-induced contraction in endothelium-denuded mouse thoracic aortas.
The concentration–response curves for OVA increased in endothelium-denuded mouse thoracic aortic rings exposed to stepwise increase in the concentration of OVA (KCl: 1.51 ± 0.18 N, Fig. 1). To evaluate the role of myosin light chain kinase (MLCK), Rho kinase, Src, EGFR, and MAPKs on OVA-induced contraction in endothelium-denuded mouse thoracic aortic rings, the aortic rings were treated with these inhibitors 15 min before exposure to OVA. The contraction of mouse thoracic aortic rings by OVA was attenuated by the Rho kinase inhibitor, Y-27632 (10 µM, p < 0.05, vs. control rings, KCl: 1.59 ± 0.21 N, Fig. 1A), the Src inhibitor, PP2 (3 µM, p < 0.05, vs. control rings, KCl: 1.54 ± 0.18 N, Fig. 1B), and the EGFR inhibitor, AG1478 (10 µM, p < 0.05, vs. control rings, KCl: 1.56 ± 0.16 N, Fig. 1B). As shown in Fig. 1C, the contractile effect of OVA was markedly blocked by the MEK inhibitor, PD98059 (10 µM, p < 0.05, vs. control rings, KCl: 1.44 ± 0.17 N), and the Erk1/2 inhibitor, FR180204 (10 µM, p < 0.05, vs. control rings, KCl: 1.49 ± 0.14 N), and partially but significantly attenuated by the JNK inhibitor, AS601245 (10 µM, p < 0.05, vs. control rings, KCl: 1.56 ± 0.17 N), and the p38 MAPK inhibitor, SB203580 (10 µM, p < 0.05, vs. control rings, KCl: 1.43 ± 0.16 N). In contrast, the MLCK inhibitor, ML-7 (10 µM, KCl: 1.49 ± 0.19 N), had no effect on OVA-induced contraction (Fig. 1A).
We reported that OVA-induced contraction depends on transactivation of EGFR via shedding of pro-HB-EGF in rat thoracic aortas.23) To investigate whether OVA-induced contraction of mouse thoracic aortas was also produced by transactivation of EGFR, we determined the effects of TAPI-0, a metalloproteinase inhibitor (10 µM), on OVA-induced contraction. The mouse thoracic aortic rings were incubated with TAPI-0 and which did not block OVA-induced contraction in endothelium-denuded mouse thoracic aortic rings (KCl: 1.57 ± 0.15 N, Fig. 1D).
Effect of OVA on Phosphorylation of MYPT1, Erk1/2, JNK, p38 MAPK, and Src in Mouse Thoracic Aortic RingsThe organ chamber experiments suggest that OVA elicits contraction of mouse thoracic aortic rings through Src, EGFR, MEK, Erk1/2, JNK, p38 MAPK, and Rho kinase. To confirm these observations, we assayed the phosphorylation levels of MYPT1 at Thr-853 as an index of Rho kinase activity,27) Erk1/2 at Thr-202/Try-204, JNK at Thr-183, p38 MAPK at Tyr-182, and Src at Tyr-416 in mouse thoracic aortic rings by Western blotting. As shown in Fig. 2, the ratio of phosphorylated protein to total protein for MYPT1, Erk1/2, JNK, p38 MAPK, and Src was increased by OVA within 2 min of exposure, and which was maintained for 10 min.
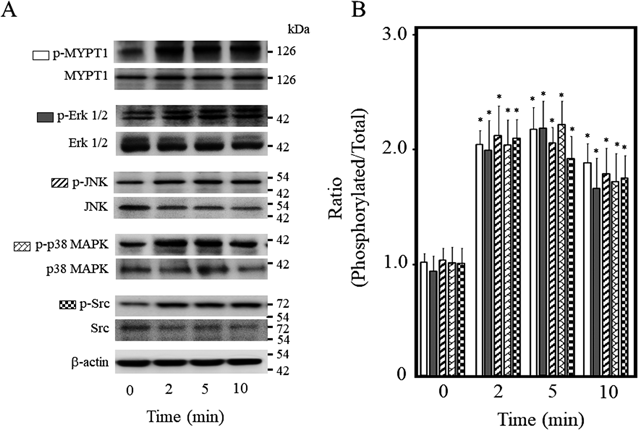
Phosphorylation levels of MYPT1 (Thr-853), Erk1/2 (Thr-202/Tyr-204), JNK (Thr-183), p38 MAPK (Tyr-182), and Src (Tyr-416) were measured in OVA (250 µM)-treated aortic rings by Western blotting. (A) Representative blots. (B) Bar graphs demonstrating densitometric quantification of the ratios of phosphorylated to total proteins. Data are means ± S.E.M. of four independent experiments, * p < 0.05 vs. control rings (0 min).
To investigate whether activation of Rho kinase is caused by the activation of Src, EGFR, MEK, Erk1/2, JNK, and/or p38 MAPK during OVA-induced contraction, we quantified the phosphorylation level of MYPT1 at Thr-853 in mouse thoracic aortic rings 5 min after OVA treatment in the presence or absence of various inhibitors (Fig. 3). OVA (250 µM) increased phosphorylation level of MYPT1, which was abrogated by Y-27632 (10 µM, p < 0.05, vs. OVA alone), AG1478(10 µM, p < 0.05, vs. OVA alone), PP2 (3 µM, p < 0.05, vs. OVA alone), PD98059 (10 µM, p < 0.05, vs. OVA alone), and FR180204 (10 µM, p < 0.05, vs. OVA alone). However, AS601245 (10 µM) and SB203580 (10 µM) had no effect on OVA-induced MYPT1 phosphorylation. These results showed that Rho kinase is a downstream target of Erk1/2, MEK, Src, and/or EGFR, which are activated by OVA in endothelium-denuded mouse thoracic aortic rings.
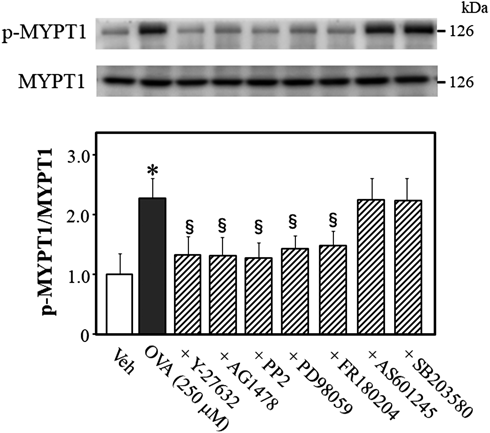
Representative blots are presented above bar graphs showing the densitometric data as the ratio of phosphorylated (Thr-853) MYPT1 to total MYPT1. Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), PD98059 (10 µM), FR180204 (10 µM), AS601245 (10 µM), SB203580 (10 µM), or vehicle (Veh; 10 µL dimethyl sulfoxide) were added 15 min before OVA (250 µM for 5 min) treatment. Data are means ± S.E.M. from 4 independent experiments, * p < 0.05 vs. Veh; § p < 0.05 vs. OVA alone.
To determine what signaling molecule regulates Erk1/2 phosphorylation while OVA induces Rho kinase activation, we quantified the phosphorylation levels of Erk1/2 at Thr-202/Try-204 in OVA-treated mouse thoracic aortic rings (Fig. 4). OVA increased Erk1/2 phosphorylation, which was blocked by AG1478 (p < 0.05, vs. OVA alone), PP2 (p < 0.05, vs. OVA alone), PD98059 (p < 0.05, vs. OVA alone), and FR180204 (p < 0.05, vs. OVA alone), but not by Y-27632, suggesting that Erk1/2 is an upstream effector of Rho kinase, and a downstream effector of EGFR, Src, and MEK.
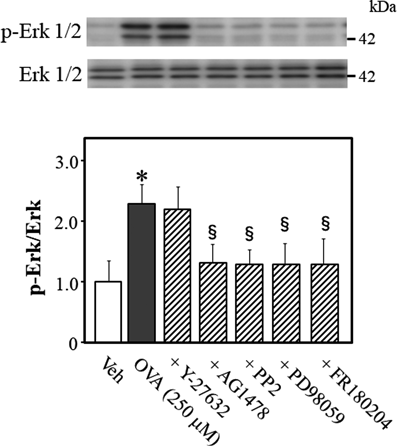
Representative blots are presented above bar graphs showing the densitometric data as the ratio of phosphorylated (Thr-202/Tyr-204) Erk1/2 to total Erk1/2. Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), PD98059 (10 µM), FR180204 (10 µM), or Veh (10 µL dimethyl sulfoxide) were added 15 min before OVA (250 µM for 5 min) treatment. Data are means ± S.E.M. from 4 independent experiments, * p < 0.05 vs. Veh; § p < 0.05 vs. OVA alone.
To determine what signaling molecule regulates JNK phosphorylation while OVA induces Rho kinase activation, we quantified the phosphorylation level of JNK at Thr-183 in OVA-treated mouse thoracic aortic rings (Fig. 5). OVA increased JNK phosphorylation, which was blocked by AG1478 (p < 0.05, vs. OVA alone), PP2 (p < 0.05, vs. OVA alone), and AS601245 (p < 0.05, vs. OVA alone), but not by Y-27632, suggesting that JNK signals are upstream of Rho kinase and downstream of EGFR and Src.
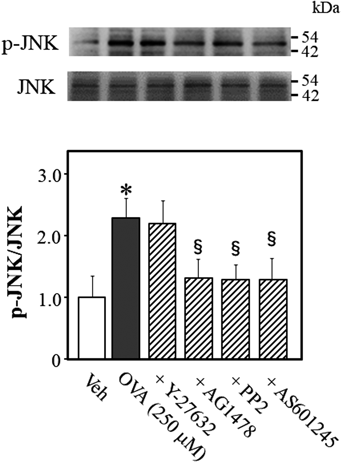
Representative blots are presented above bar graphs showing the densitometric data as the ratio of phosphorylated (Thr-183) JNK to total JNK. Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), AS601245 (10 µM), or Veh (10 µL dimethyl sulfoxide) were added 15 min before OVA (250 µM for 5 min) treatment. Data are means ± S.E.M. from 4 independent experiments, * p < 0.05 vs. Veh; § p < 0.05 vs. OVA alone.
To determine what signaling molecule regulates p38 MAPK phosphorylation while OVA induces Rho kinase activation, we quantified the phosphorylation level of p38 MAPK at Tyr-182 in OVA-treated mouse thoracic aortic rings (Fig. 6). OVA increased p38 MAPK phosphorylation, which was abolished by AG1478 (p < 0.05, vs. OVA alone), PP2 (p < 0.05, vs. OVA alone), and SB203580 (p < 0.05, vs. OVA alone), but not by Y-27632, suggesting that p38 MAPK signals are upstream of Rho kinase and downstream of EGFR and Src.
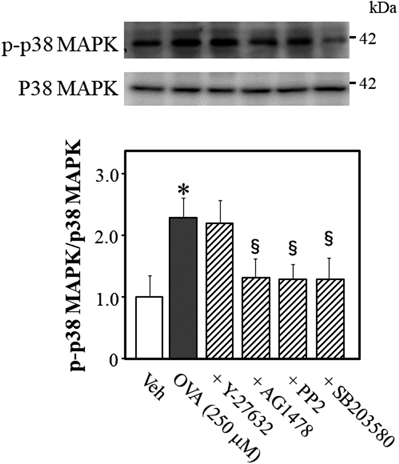
Representative blots are presented above bar graphs showing the densitometric data as the ratio of phosphorylated (Tyr-182) p38 MAPK to total p38 MAPK. Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), SB203580 (10 µM), or Veh (10 µL dimethyl sulfoxide) were added 15 min before OVA (250 µM for 5 min) treatment. Data are means ± S.E.M. from 4 independent experiments, * p < 0.05 vs. Veh; § p < 0.05 vs. OVA alone.
To determine what signaling molecule regulates Src phosphorylation while OVA induces Rho kinase activation, we quantified the phosphorylation level of Src at Tyr-416 in OVA-treated mouse thoracic aortic rings (Fig. 7). OVA increased Src phosphorylation, which was prevented by PP2 (p < 0.05, vs. OVA alone), but not Y-27632, AG1478, PD98059, FR180204, AS601245, or SB203580, suggesting that Src signals are upstream of Rho kinase, EGFR, MEK, Erk1/2, JNK, and p38 MAPK.
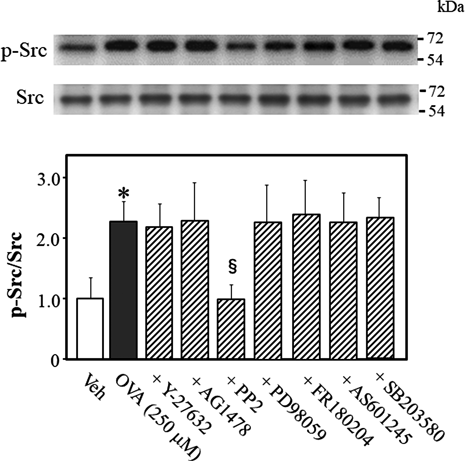
Representative blots are presented above bar graphs showing the densitometric data as the ratio of phosphorylated (Tyr-416) Src to total Src. Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), PD98059 (10 µM), FR180204 (10 µM), AS601245 (10 µM), SB203580 (10 µM), or Veh (10 µL dimethyl sulfoxide) were added 15 min before OVA (250 µM for 5 min) treatment. Data are means ± S.E.M. from 4 independent experiments, * p < 0.05 vs. Veh; § p < 0.05 vs. OVA alone.
We reported that Src is the upstream effector of EGFR during OVA-induced Rho kinase activation in rat thoracic aortic and mesenteric arterial rings.22,23) Therefore, we determine whether Src is the upstream effector of EGFR while OVA elicits Rho kinase activation in mouse thoracic aortic rings by measurement of phosphorylation level of EGFR at Try-1173 in OVA-treated mouse thoracic aortic rings (Fig. 8). OVA increased EGFR phosphorylation, which was prevented by PP2 (p < 0.05, vs. OVA alone). AG1478 tended to inhibit OVA-induced EGFR phosphorylation (p = 0.1972, vs. OVA alone). These data suggested that Src signals are upstream of EGFR.
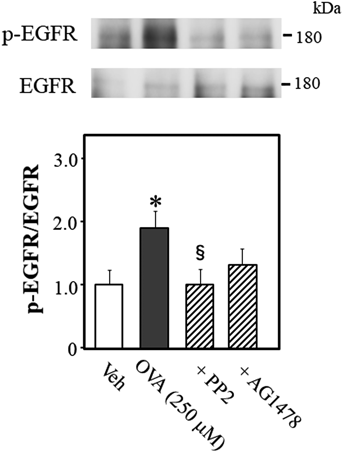
Representative blots are presented above bar graphs showing the densitometric data as the ratio of phosphorylated (Tyr-1173) EGFR to total EGFR. AG1478 (10 µM), PP2 (3 µM), or Veh (10 µL dimethyl sulfoxide) were added 15 min before OVA (250 µM for 5 min) treatment. Data are means ± S.E.M. from 5 independent experiments, * p < 0.05 vs. Veh; § p < 0.05 vs. OVA alone.
The principal finding of this study was that OVA produced contractile effects in endothelium-denuded thoracic aortas in mice. Contraction of mouse thoracic aortic rings is mediated by Src, EGFR, MEK, Erk1/2, p38 MAPK, JNK, and Rho kinase-dependent signaling. Phosphorylation of MYPT1, which was stimulated by OVA, was regulated by Src, EGFR, MEK, Erk1/2, and/or Rho kinase, because inhibition of these proteins abolished MYPT1 phosphorylation. To our knowledge, this is the first report on the contractile effects of OVA in mice.
The contraction of vascular smooth muscle is primarily regulated by the Ca2+-dependent activation of MLCK, which phosphorylated MLC.28) The phosphorylated MLC is dephosphorylated by MLCP which consisted with three units: a 37-kDa catalytic subunit (PP-1C), a 110- to 130-kDa regulatory subunit (MYPT1), and a 20-kDa subunit.29) Phosphorylation of MYPT1 by Rho kinase leads to inactivation of MLCP, which induced Ca2+ sensitization, thereby enhancing contraction without increasing concentration of Ca2+ or MLCK activity.30) Mori and Tsushima reported that OVA induces contraction of guinea pig ileal longitudinal smooth muscle through Rho kinase, because the Rho kinase inhibitor abolished OVA-induced contraction.21) We also reported that OVA-induced contraction was regulated by Rho kinase in rat thoracic aortas and mesenteric arteries.22,23) However, it is unknown whether OVA induces contraction in mouse thoracic aortas through the similar contraction mechanism of rat thoracic aortas and mesenteric arteries. Here, we showed that the primary role for Rho kinase in mouse thoracic aortas on OVA-induced contraction, because the Rho kinase inhibitor, Y-27632, reduced OVA-induced contraction and MYPT1 phosphorylation, whereas the MLCK inhibitor, ML-7, had no effect on OVA-induced contraction. This is consistent with previous findings that the OVA-induced contractile effect on smooth muscle cells was not accompanied by a rise in cytosolic-free Ca2+, but instead occurred as a result of increased Ca2+ sensitivity of the contractile apparatus.16,31,32)
The binding of EGF to EGFR induces dimerization of the EGFR. The dimerization of EGFR results in the autophosphorylation of five tyrosine residues (Tyr 1173, 1148, 1086, 1068, and 992) in the intracellular site of EGFR. The contraction of vascular smooth muscle by EGF was observed in rat thoracic aortas of hypertensive models,33,34) but not normotensive rats.35) In addition, EGF-induced contraction was regulated by Erk1/2 and Rho kinase in DOCA-salt hypertensive rats.35) These reports suggested that EGF induced smooth muscle contraction in hypertensive rats, but not normotensive rats, was regulated by Erk1/2 and Rho kinase. We also showed that EGF did not induce the contraction of endothelium-denuded thoracic aortas in normotensive rats.24) However, in this study, OVA-induced contraction and MYPT1 phosphorylation were abolished by the inhibitor of EGFR. We reported that EGF enhanced OVA-induced contraction of endothelium-denuded thoracic aortas in normotensive rats, which was abolished by EGFR, Erk1/2, and Rho kinase inhibitors.24) This evidence indicates that signaling cascades of EGFR have important role for contraction by OVA.
Src was shown to mediate phosphorylation of EGFR at Tyr-845 in the cytoplasm36,37) or indirectly via the metalloproteinase-catalyzed formation of HB-EGF from pro-HB-EGF.38) In a our previous study, we showed that OVA-induced contraction was mediated by indirect activation of EGFR via shedding of pro-HB-EGF in rat thoracic aortas.23) In contrast, OVA-induced contraction of rat mesenteric arterial rings was not mediated by indirect activation of EGFR, because a metalloproteinase inhibitor, TAPI-0, and a diphtheria toxin mutant, CRM 197, that specifically blocks the action of pro-HB-EGF, had no effect on this contraction.22) In this study, we showed that OVA-induced contraction of mouse thoracic aortic rings was blocked by inhibitors of EGFR and Src, but not by TAPI-0, and OVA-induced EGFR phosphorylation was blocked by PP2, suggesting that, in mouse thoracic aortas, OVA-induced contraction is mediated by direct activation of EGFR in common with rat mesenteric arteries.22) In rat thoracic aortas, we showed that OVA-induced contraction depended on indirectly activation of EGFR.23) The reasons for the differences in OVA-induced contraction in mouse and rat thoracic aortas are not clear, and further studies are needed to find the mechanisms underlying the differences in EGFR activation by Src which was activated by OVA in different animal species.
MAPKs play an important role in intracellular signaling initiated by various stimuli. We previously showed that EGF increased Ca2+ sensitization in rat thoracic aortic rings and stimulated phosphorylation of Erk1/2 and MYPT1 in rat vascular smooth muscle cells.24) In addition, co-stimulation with EGF- and OVA-induced contraction in rat thoracic aortic rings or OVA-induced contraction in rat mesenteric arteries was mediated by Erk1/2 and MEK, because contraction and MYPT1 phosphorylation were abolished by the treatment with Erk1/2 or MEK inhibitor.22,26) In present study, we showed that OVA-induced contraction in mouse thoracic aortas was strongly inhibited by Erk1/2 or MEK inhibitor, and partially but significantly inhibited by JNK or p38 MAPK inhibitor. Furthermore, OVA treatment of mouse thoracic aortic rings facilitated phosphorylation of Erk1/2, JNK, and p38 MAPK, and these kinases are downstream of Src and EGFR. On the other hand, OVA-induced phosphorylation of MYPT1 was inhibited by Erk1/2 or MEK inhibitor, but not by JNK or p38 MAPK inhibitor. These results indicate that Erk1/2 and MEK mainly regulate OVA-induced contraction and phosphorylation of MYPT1, whereas JNK and p38 MAPK are partially involved in OVA-induced contraction through pathways independent of MYPT1 phosphorylation. We previously reported that OVA-induced contraction in rat mesenteric arteries or EGF- and OVA- induced contraction in rat thoracic aortas was strongly inhibited by Erk1/2 or MEK inhibitor, but not by JNK or p38 MAPK inhibitor. The reasons for the differences between mouse and rat arteries in the involvement of MAPKs in OVA-induced contraction are unclear. In mouse thoracic aortas, we showed that JNK and p38 MAPK are partially involved in OVA-induced contraction via pathways independent of MYPT1 activation. There is evidence that JNK and p38 MAPK increase cofilin phosphorylation in endothelial cells.39) Cofilin is inactivated by phosphorylation, resulting in polymerization and stabilization of actin filaments,40–42) suggesting that JNK and p38 MAPK regulate OVA-induced contraction through actin polymerization via cofilin inactivation in mouse thoracic aortas. However, the mechanism of contraction in mouse thoracic aortas involving JNK and p38 MAPK is not clear. Further studies are needed to explore the mechanism underlying this contraction via JNK and p38 MAPK.
In summary, the present study provides novel findings that OVA induces contraction in mouse thoracic aortas. OVA-induced contraction in mouse thoracic aortas was modulated by EGFR, Src, MAPKs, and Rho kinase. Vanadates inhibit unidentified PTPase(s), which in turn causes Src and EGFR activation. EGFR induces activation of MEK, Erk1/2, and Rho kinase, which leads to inactivation of MLCP via MYPT1 phosphorylation and increases mouse thoracic aortic contractility. OVA-induced contraction in mouse thoracic aortic rings is not indirect transactivation of EGFR, but only direct activation of EGFR. However, the reasons for the difference in OVA-induced contraction between mouse thoracic aortas and rat thoracic aortas are not fully understood. Moreover, we could not identify the target of OVA in Src-dependent constriction. Further studies are needed to comprehensively evaluate the regulation of PTPases in order to understand the mechanisms underlying contraction. The function of vascular smooth muscle is regulated by the balance of protein tyrosine kinases and protein tyrosine phosphatases. Disruption of the cellular tyrosine phosphorylation equilibrium causes several diseases such as diabetes and hypertension.43–45) Therefore, treatment of OVA mimics such conditions, and our present study provide evidence regarding contractile effects of OVA in mice, and has important implications in vascular physiology and disease.
This study was partially supported by a Grant-in-Aid for Scientific Research (C) (No. 15K07984) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
The authors declare no conflict of interest.