Abstract
Prostate cancer is the second most common cancer among men worldwide, and it is ranked first in the United States and Europe. Since prostate cancer is slow-growing, active surveillance for low-risk cancer has been increasingly supported by various guidelines. Most prostate cancers reactivate telomerase to circumvent the replicative senescence caused by the end replication problem; therefore, telomerase inhibition is potentially useful for the suppression of prostate cancer progression during this active surveillance or for the prevention of cancer recurrence after conventional therapies. In this study, we demonstrated that the perylene derivatives, PM2 and PIPER, could suppress human telomerase reverse transcriptase (hTERT) expression and telomerase activity in the short-term treatment of androgen-dependent prostate cancer cell line LNCaP and the androgen-independent prostate cancer cell line PC3 prostate cancer cells. Long-term treatment with subcytotoxic doses of these compounds in both prostate cancer cells showed telomere shortening and a significant increase in senescent cells. Although the acute cytotoxicity of PM2 was about 30 times higher than that of PIPER in both prostate cancer cells, the cellular uptake of both compounds was comparable as determined by flow cytometry and fluorescent microscopy.
INTRODUCTION
Prostate cancer is the second most common cancer among men worldwide, and it is ranked first in the United States and Europe.1–3) In Asia, the incidence of prostate cancer is relatively low, but there has been a rapid rise of prostate cancer incidence due to the introduction of prostate-specific antigen (PSA) testing.3) Although the PSA-based screening was found to correlate with mortality reduction, it also coincided with high proportions of unnecessary biopsies, overdiagnosis, and overtreatment.3) Compared with other types of cancers, prostate cancer grows relatively slowly and sometimes causes no problems for years.4,5) Treatment with surgery or radiation is standard local therapy, but about one-third of patients will develop biochemical recurrence,6) and many are living with urinary or sexual function problems afterward.7) As a result, active surveillance is now recognized as a preferred strategy in patients with low-risk localized prostate cancer as an alternative to the immediate radical treatment.6,8) For patients with metastatic prostate cancer, androgen deprivation therapy (ADT) is the standard of care. However, most patients progress to castration-resistant prostate cancer (CRPC).6,9) Considering the advanced age of most patients, the current chemotherapeutic drugs have done little to improve the survival rate of these CRPC patients.10) Therefore, new targeted therapies with fewer side effects are urgently needed for the management of prostate cancer, both in the active surveillance state and in the metastatic CRPC. One such strategy, telomerase inhibition after conventional therapeutic approaches (surgery and chemotherapy/radiotherapy), was suggested to be an ideal strategy for prostate cancer therapy.11)
Progressive telomere shortening from cell division provides a barrier for cancer progression.12) With each round of cell division, telomeric DNA is shortened by 50–200 base pairs due to the end replication problem.13) The cells are allowed to replicate for a number of cell divisions until a few telomeres are shortened to a critical length, at which the shortening triggers an irreversible cell cycle arrest called replicative senescence.14) Most cancer cells, including prostate cancers, reactivate telomerase to maintain their telomeres.3,15) Human telomerase is a multi-subunit ribonucleoprotein complex in which the isolated catalytically active enzyme consists of two molecules each of human telomeric RNA (hTR), human telomerase reverse transcriptase (hTERT), and dyskerin.16) Human telomerase uses its internal RNA as a template to catalyze the addition of 6-base pairs repeats (TTA GGG)n to the 3′ telomere ends, and thereby prevents the telomeres from becoming critically short.17) Although hTR and hTERT are both essential components for telomerase activity and telomere maintenance, transcriptional regulation of hTERT is the predominant mechanism for controlling telomerase activity.18)
Among several strategies to inhibit telomerase, those based on G-quadruplex ligands are perhaps the most widely studied.19) G-Quadruplex ligands prevent the access of telomerase to the 3′ telomeric ends by facilitating and/or stabilizing G-quadruplex formation of G-rich telomeric DNA.18) They are also found to down-regulate hTERT expression by facilitating the formation of G-quadruplex at the hTERT promoter, preventing the transcriptional machinery from accessing its promoter.20) In the same manner, G-quadruplex ligands have been shown to stabilize the G-quadruplex structure on the G-quadruplex motif from the promoters of several genes controlling cellular proliferation, such as c-MYC, c-KIT, k-RAS, and vascular endothelial growth factor (VEGF), among others, leading to the down-regulation of these genes and antiproliferative activity in cancer cell lines.21) Moreover, G-quadruplex ligands were also found to facilitate G-quadruplex formation at the androgen receptor (AR) promoter, leading to the down-regulation of AR gene expression in AR-positive prostate cancer cells.22,23)
Several perylene derivatives have been found to be G-quadruplex ligands and telomerase inhibitors.24,25) Most of these perylene derivatives are perylene diimide derivatives, which are derived from the first perylene-based G-quadruplex ligand and telomerase inhibitor, PIPER26) (Fig. 1). Since then, PIPER has been often used as a standard compound to compare with other perylene derivatives. In our laboratory, we have been particularly interested in a perylene monoimide derivative, PM2 (Fig. 1). This compound is more water soluble than PIPER and has asymmetric side chains that might cause this compound to act differently than PIPER or other perylenediimide derivatives. Previously, we reported that PM2 and PIPER facilitated the G-quadruplex formation of the telomeric DNA sequence and the hTERT promoter DNA sequence, leading to the inhibition of telomerase activity and suppression of hTERT expression in A549 lung cancer cells, respectively.27) The long-term treatment of A549 cells with a subcytotoxic dose of both perylene derivatives led to telomere shortening, decreased cell proliferation and tumorigenicity, and cellular senescence. As mentioned earlier, prostate cancer is a slow-growing cancer, and patients with this type of cancer are mostly advanced in age. This makes prostate cancer a good candidate for telomerase inhibition therapy during active surveillance or after conventional therapeutic approaches, due to its non-cytotoxic target-oriented strategy. And since the nature of each cancer is different, particularly in this case, the telomere length, we examined the efficacy of PM2 and PIPER on hTERT expression, telomerase inhibition, telomere length attrition, and cellular senescence of the two most commonly studied prostate cancer cell lines, the androgen-dependent prostate cancer cell line LNCaP and the androgen-independent prostate cancer cell line PC3 in this study. Furthermore, we also investigated the effect of these two compounds on transcriptional repression of the AR receptor, which would have an additional benefit in prostate cancer therapy. Because PM2 and PIPER differ in their states of acute cytotoxicity, we also investigated their partition coefficients and cellular uptake in these two prostate cancer cell lines.
MATERIALS AND METHODS
Chemicals and ReagentsSamples of PM2 and PIPER were synthesized using previously published protocols.28,29) The characterization data for both compounds are consistent with the literature and are provided in the Supplementary Materials Figs. S1 and S2. The absorption spectra and the fluorescence emission spectra are provided in Supplementary Material Fig. S3. All other chemicals were of molecular biology grade and purchased from commercial suppliers. All oligonucleotides and fluorescence-tagged oligonucleotides were purchased from Ward Medic (Thailand).
Cell CultureHuman prostate cancer cell lines LNCaP and PC3 were obtained from the American Type Culture Collection (Rockville, MD, U.S.A.). The cells were cultured in Roswell Park Memorial Institute medium 1640 (RPMI 1640) with 10% fetal bovine serum (FBS) and 1% antibiotics (50 units/mL penicillin, 50 µg/mL streptomycin) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The peripheral blood mononuclear cells (PBMC) were collected from healthy volunteers and cultured in the same medium and conditions.
Cell Growth Inhibition AssayThe cell growth inhibition of the perylene derivatives was determined using the sulforhodamine B (SRB) assay according to the published protocol.30) The indicated cancer cells (1.0 × 104 cells) or PBMC cells (1.0 × 105 cells) were incubated with various concentrations of either PM2 or PIPER at 37°C for 72 h in a humidified incubator with 5% CO2. The IC50 was calculated from the dose–response relationship curve between the drug concentration and the percentage of cell viability using the software CurveExpert 1.4. The reported results represent the mean values of three independent experiments.
Semi-quantitative RT-PCR AnalysisProstate cancer cells (LNCaP or PC3, 5.0 × 105 cells) were grown on a 6-well tissue culture plate for 24 h before being treated with various concentrations of PM2 or PIPER for 24 h at 37°C in a humidified CO2 (5%) incubator. The total RNA was collected, and the mRNA was converted into cDNA using oligo-(dT)18 primer and RevertAid reverse transcriptase (Thermo Scientific, U.S.A.) according to the manufacturer's instructions. The cDNAs were then amplified by PCR using specific primers for each gene. Each PCR cycle was carefully chosen so that the intensity of the detected PCR product was proportional to the initial amount of cDNA in the reaction (see Supplementary Material Fig. S4). PCR products were then separated by agarose gel electrophoresis and visualized under UV light using Nucleic Acid Staining solution (RedSafe™, Intron Biotechnology, U.S.A.). The primer sequences, annealing temperatures, and PCR cycles are summarized in the Supplementary Material Table S1.
Modified Fluorescent Telomeric Repeat Amplification Protocol (TRAP) AssayProstate cancer cells (LNCaP or PC3, 5.0 × 105 cells) were grown on a 6-well tissue culture plate for 24 h before being treated with various concentrations of PM2 or PIPER for 48 h at 37°C in a humidified CO2 (5%) incubator. The cells were lysed with 50 µL of CHAPS lysis buffer [10 mM Tris–HCl (pH 7.5), 1 mM MgCl2, 0.1 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 5 mM β-mercaptoethanol, 0.5% CHAPS, 10% glycerol, protease inhibitor cocktail, and 200 units/mL RNase inhibitor] and centrifuged at 12000 × g for 10 min. The supernatant was collected and quantified for protein concentration by Bradford assay (BioRad, U.S.A.). A 40 µg sample of protein from this supernatant served as a source of telomerase for the TRAP assay. A modified fluorescent TRAP assay was performed according to a published protocol; this assay uses a specific primer to prevent the shortening and lengthening of the original telomerase products as normally seen from a standard TRAP assay.31) Briefly, the 40 µg sample of crude telomerase from the cells treated with the test sample were incubated with 35 µL of telomerase reaction buffer [20 mM Tris–HCl (pH 8.3), 1.5 mM MgCl2, 63 mM KCl, 1 mM EGTA, 0.1 mg/mL bovine serum albumin, 0.005% Tween 20, 200 µM deoxyribonucleoside triphosphates (dNTPs), and 15 pmol MTS primer] at 30°C for 30 min. The telomerase-extended products were then amplified by adding 15 µL of amplification reaction mixture (2.5 units Taq DNA polymerase, 15 pmol RP-FAM primer, 0.25 pmol RPc3g, 0.01 amol IC, and 7.5 pmol NT primer in 20 mM Tris–HCl, pH 8.3), and PCR was performed in a thermocycler with the following conditions: 3 cycles of (95°C for 30 s; 58°C for 60 s; and 72°C for 90 s) and 28 cycles of (95°C for 30 s; 65°C for 30 s; and 72°C for 30 s). The amplification products were separated by non-denaturing acrylamide gel electrophoresis and visualized with a phosphoimaging system (Typhoon; Molecular Dynamics). The oligonucleotides used in this assay are summarized in Supplementary Material Table S2.
Long-Term Proliferation AssayFor the long-term proliferation assay, three sets of cell cultures were compared: the control group and the two experimental groups with the subcytotoxic doses of a test compound added to the culture media. The LNCaP cells (8 × 105 cells) or PC3 cells (2 × 105 cells) were first seeded onto a 75 cm2 tissue culture flask in RPMI 1640 medium supplemented with 10% fetal bovine serum, with or without the indicated concentration of PM2 or PIPER. The culture media in each set was then changed after 3 d. After the cells reached confluence on Day 6, the cells were trypsinized and counted. The same number of cells for each prostate cancer were then subcultured onto a new 75 cm2 tissue culture flask, and the process was repeated for up to 90 d. The remaining cells in each passage were collected and used for telomere length assay and senescence-associated β-galactosidase activity assay. The number of population doubling was calculated by the equation: n = (log Pn − log P0)/log 2, where Pn is the number of cells after n doublings and P0 is the initial seeding density. The cumulative number of population doubling was then plotted against time.
Telomere Length AssayThe average telomere length of LNCaP and PC3 cells, collected at the indicated passages from the long-term proliferation assay, were assayed using the TeloTAGGG Telomere Length Assay kit (Roche Applied Science, Germany) according to the manufacturer’s instructions. In brief, total DNA was isolated from the prostate cancer cells using DNAzol® (Invitrogen, U.S.A.). Then, 8 µg of purified genomic DNA was digested by two restriction enzymes (HinfI and RsaI), before the digested DNA fragments were separated by 0.8% agarose gel electrophoresis and blotted onto a nylon membrane (Immobilon™-Ny+, Millipore, U.S.A.). The telomeric DNA fragments were then hybridized with a DIG-labeled telomeric probe, followed by DIG-specific antibody coupled with alkaline phosphatase, and visualized on X-ray film using a chemiluminescent system. The average telomere restriction fragment (TRF) length was calculated according to the formula ∑(ODi)/∑(ODi/Li), where ODi indicates the chemiluminescent signal at the position i, and Li is the molecular weight marker at the same position.
Senescence-Associated β-Galactosidase Activity AssayLNCaP and PC3 cells (1 × 103 cells), collected at the indicated passages from the long-term proliferation assay, were seeded on glass cover slides and grown under 500 µL of culture media for 24 h. The cells were washed twice with phosphate buffered saline (PBS) and fixed with 2% formaldehyde and 0.2% glutaraldehyde solution for 5 min at room temperature. The cells were then washed twice with PBS and incubated with the senescence-associated β-galactosidase (SA-β gal) solution (1 mg/mL X-gal, 40 mM citric acid/sodium phosphate (pH 6), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2) for 24 h at 37°C. The X-gal solution was removed, and the cells were rinsed once with PBS. The β-galactosidase positive cells were monitored under a phase contrast microscope with a blue stain, and were usually accompanied with cell morphological changes. The percentage of β-galactosidase positive cells was calculated and plotted against time.
Octanol/H2O Partition Coefficient (Pow) by Shake Flask MethodThe general procedure of this experiment was based on OECD Test Guideline 107.32) The calculation of Pow was based on absorbance measurements of a single liquid phase previously described by Wattanasin et al.33) Measurements of Pow for PM2 and PIPER were performed using 1-octanol (Fluka, Switzerland) and 10 mM Tris buffer (pH 10) as solvents. Measurements of Pow for caffeine and riboflavin as reference compounds were also performed using 1-octanol and 10 mM PBS buffer (pH 7.4) as solvents.
Cellular Uptake of PM2 and PIPER by Flow CytometryLNCaP and PC3 cells (5 × 105 cells) were seeded on a 6-well plate for 24 h before they were treated with the indicated concentrations of PM2 or PIPER for 24 h at 37°C in a humidified 5% CO2 incubator. The cells were then washed, and the trypsinized cells were collected by centrifuging at 500 × g for 5 min. The cells were resuspended in 500 mL PBS before they were analyzed using CyAn ADP flow cytometer equipped with Kaluza, Flow Cytometry Analysis Software (Beckman Coulter, U.S.A.). The data were collected from 50000 gated events with λEx of 488 nm and λEm of 680 nm.
Fluorescence MicroscopyLNCaP or PC3 cells (1 × 105 cells) were seeded on a glass cover slide under 500 µL of culture media for 24 h. The cells were then washed twice with PBS and treated with 2 mL of the indicated concentration of a perylene derivative in culture media for 48 h before the cell nuclei were stained with 500 µL of 100 nM 4′,6-diamidino-2-phenylindole (DAPI) for 45 min. The excess dye was then washed before the intracellular localization of DAPI (using a DAPI filter) and the perylene derivative (using a Red filter) were accessed using an Olympus AX70 fluorescence microscope (Olympus, Japan).
Statistical AnalysisAll values are given as mean ± standard derivation (S.D.) from triplicate samples of three independent experiments. The Student’s t-test and two-way ANOVA with SPSS 11.5 software package were used to compare the treated and control cells. Differences are considered statistically significant when p < 0.05 or < 0.01.
RESULTS
Acute Cytotoxicity of PM2 and PIPER in Prostate Cancer Cell Lines and PBMC CellsThe sulforhodamine B (SRB) assay was employed to evaluate the acute cytotoxicity of PM2 and PIPER in prostate cancer cell lines (LNCaP and PC3) and PBMC. The prostate cancer cells and PBMC were incubated with various concentrations of either PM2 or PIPER for three days. The IC50 was calculated from the dose-response relationship curve between the drug concentration and the percentage of cell viability using the software CurveExpert 1.4 (see Supplementary Material Fig. S5). The IC50 values for PM2 in LNCaP and PC3 cells were 3.0 ± 0.3 and 3.2 ± 1.0 µM, respectively, while the IC50 values for PIPER in LNCaP and PC3 were 89.6 ± 16.3 and 92.7 ± 8.5 µM, respectively. The cytotoxicity of PM2 and PIPER is similar to our published results in A549 lung cancer cells, in which the IC50 of PM2 and PIPER were 4.0 ± 0.1 and 52.4 ± 0.2 µM, respectively.27) As indicated by these results, PM2 is much more toxic in cancer cells than PIPER. Both PM2 and PIPER appear to have little effect on peripheral blood mononuclear cells, in which the percentage of cell viability remained relatively constant in the presence of 0–160 µM of either PM2 or PIPER (see Supplementary Material Fig. S5C).
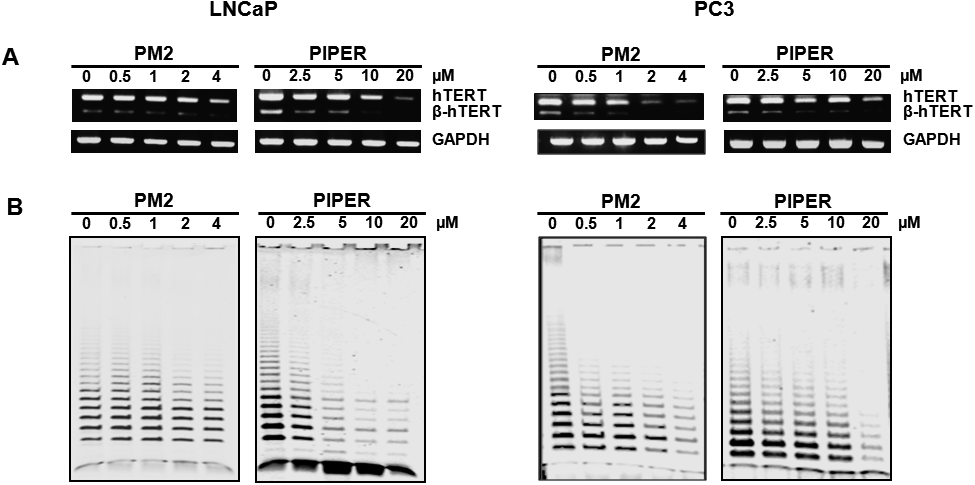
Suppression of hTERT Expression and Telomerase Activity by PM2 and PIPER in LNCaP and PC3 Prostate Cancer CellsPreviously, PM2 was found to be more effective than PIPER in terms of G-quadruplex formation, down-regulation of hTERT expression, and suppression of telomerase activity in A549 lung cancer cells.27) To evaluate these abilities in LNCaP and PC3 prostate cancer cells, we employed the same semi-quantitative RT-PCR to assess mRNA gene expression and modified TRAP assay to assess telomerase activity. For the RT-PCR assay, the prostate cancer cells were incubated with 0–4 µM of PM2 or 0–20 µM of PIPER for 24 h before the total mRNAs were converted to cDNAs and amplified using gene-specific primers. As illustrated in Fig. 2A, both PM2 and PIPER suppressed hTERT gene expression in a concentration-dependent manner in both LNCaP and PC3 cells, while the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene was not affected. For the TRAP assay, the prostate cancer cells were incubated with 0–4 µM of PM2 or 0–20 µM of PIPER for 48 h before crude protein extract was used as the source of telomerase in the TRAP assay. As illustrated in Fig. 2B, both PM2 and PIPER suppressed telomerase activity in a concentration-dependent manner in both LNCaP and PC3 cells. Telomerase activity was markedly reduced at the lowest concentration of PM2 and PIPER used, and PM2 appeared to suppress telomerase better than PIPER for both types of prostate cancer cells.
As mentioned above, G-quadruplex ligands have been found to facilitate G-quadruplex formation at the promoter sequences from several cancer-related genes and androgen receptor (AR). Our previous publications have also demonstrated that PM2 and PIPER induced G-quadruplex formation using short DNA sequences from the hTERT and VEGF promoters, and the expression of these genes in A549 lung cancer cells was suppressed.27,34) In this study, the gene expressions of hTERT, c-Myc, and VEGF from both LNCaP and PC3 cells treated with either PM2 or PIPER were found to be down-regulated, while the gene expressions of other telomerase-related genes: hTR, TRF1, TRF2, and hTEP1, which have no G-quadruplex motif on their promoters, were not affected (see Supplementary Material Fig. S6). The gene expression of AR, as well as the AR-signaling downstream prostate-specific antigen (PSA) gene, in LNCaP cells were also suppressed at both mRNA and protein levels by PM2 and PIPER (see Supplementary Material Fig. S7. We did not perform these experiments in PC3 cells because these cells do not express AR). Therefore, the suppression of hTERT gene expression in these prostate cancer cells is likely due to the obstruction of the transcription machinery by G-quadruplex formation at its promoter.
Effects of Long-Term Treatment with Subcytotoxic Doses of PM2 and PIPER in LNCaP and PC3 CellsMost prostate cancer cells, including LNCaP and PC3 cells, maintain their telomere length by reactivating telomerase.35) Therefore, treatment with a telomerase suppressor should allow these cancer cells to exhibit telomere shortening after successive rounds of cell division in the same manner as normal somatic cells, albeit faster due to the rapid cell division in cancer cells. When one or more telomeres are shortened to a critical length, the cell is triggered to enter an irreversible cell cycle arrest called cellular senescence.13) Ideally, a specific telomerase inhibitor should cause telomere shortening in cancer cells without interfering with other cellular mechanisms, which might otherwise affect normal somatic cells and thereby cause side effects. Our previous study showed that PM2 and PIPER could directly inhibit telomerase in an in vitro TRAP assay,27) and in the present study, we showed that they could suppress hTERT gene expression and telomerase activity in both LNCaP and PC3 prostate cancer cells. As such, our next step was to investigate whether subcytotoxic doses of these two compounds would allow the LNCaP and PC3 cells to proliferate for several generations and display telomere shortening and subsequent cellular senescence.
To this end, LNCaP and PC3 cells were treated with 0.4 and 0.8 µM of PM2, or 1.0 and 2.0 µM of PIPER, supplemented in the culture media, with the change of fresh media every 3 d and subculturing every 6 d. The cells were counted, and the numbers of population doublings were calculated. The graph plotted between the cumulative number of population doublings and incubation time is shown in Fig. 3. The plots were linear in the control set for both LNCaP cells (R2 = 0.9996) and PC3 cells (R2 = 0.9995). Based on the graph, the population doubling time (PDT) of the LNCaP cells in the control set was 3.8 d, while the PDT of the PC3 cells was 2.4 d. The PDT of the PC3 cells, the more aggressive form of prostate cancer cells, was about 1.6-fold faster than that of the LNCaP cells. The presence of subcytotoxic doses of PM2 or PIPER decreased the population doubling slightly in a concentration-dependent manner (Figs. 3A–D). The effect on the population doubling of both compounds was minimal in the early rounds of cell passage, but it declined at a higher rate during the latter rounds of cell passage. In particular, the LNCaP cells treated with 0.8 µM of PM2 or 2.0 µM of PIPER proliferated so much slower after day 60 that we were unable to harvest sufficient cells to perform our subsequent experiments.
To investigate whether this decline in population doubling by PM2 and PIPER was due to cellular senescence, we collected LNCaP and PC3 cells on the indicated day to test for β-galactosidase activity, a common senescence marker. The β-galactosidase positive cells were monitored under a phase-contrast microscope with a blue stain, usually accompanied with cell morphological changes. The percentage of β-galactosidase positive cells (% β-gal cells) was then calculated and plotted against time. Figures 3E and F show that on day 6, the % β-gal cells of both LNCaP and PC3 treated with PM2 or PIPER were not substantially different from the controls, suggesting that neither compound induced cellular senescence during this short-term treatment. However, the fraction of % β-gal cells increased significantly in a time- and concentration-dependent manner. The % β-gal cells in LNCaP and PC3 cells treated with PM2 or PIPER collected at day 36 were under 20%. In contrast, at day 60, the % β-gal cells in LNCaP treated with 0.4 and 0.8 µM of PM2, 1.0 and 2.0 µM of PIPER, were up to 34, 35, 32, and 38%, respectively; while the % β-gal cells of PC3 cells collected at day 90 were 38, 48, 49, and 56%, respectively. From these data, we conclude that the decline in population doubling in both LNCaP and PC3 cells in the long-term treatment with PM2 and PIPER is likely caused by cellular senescence in these cells.
Next, we investigated whether the increase in cellular senescence in the long-term treatment of LNCaP and PC3 cells with PM2 and PIPER correlated with telomere shortening in these cells. The LNCaP cells collected on days 6, 36, and 60, and the PC3 cells harvested on day 6, 36, and 90, were subjected to the telomere length analysis. The genomic DNAs extracted from the cells were first digested with HinfI and RsaI before the telomere restriction fragments (TRFs) were analyzed by Southern blotting using a TeloTAGGG Telomere Length Assay kit. As illustrated in Fig. 4A, the mean TRF lengths of LNCaP cells in the control set remained relatively constant at around 2.6 kb over a course of 60 d. In the cells treated with 0.4 and 0.8 µM of PM2, the mean TRF length decreased slightly with time, while the difference between the two doses was insubstantial. The mean TRF lengths in the LNCaP cells treated with 1.0 and 2.0 µM of PIPER also decreased progressively with time, but the decrease was more discernible in the cells treated with 2.0 µM of PIPER.
In the experiments with PC3 cells, as demonstrated in Fig. 4B, the mean TRF lengths of PC3 cells in the control set were in the range of 8.5–8.7 kb during the course of 90 d. However, the mean TRF lengths in the cells treated with either PM2 or PIPER noticeably decreased progressively with time. The mean TRF lengths decreased from 8.6 kb on day 6, to 7.9 kb on day 36, and to 6.8 kb on day 90 in the PC3 cells treated with 0.4 µM of PM2; while in the cells treated with 0.8 µM of PM2, the mean TRF lengths decreased from 8.7 kb on day 6, to 7.7 kb on day 36, and to 6.2 kb on day 90. In the PC3 cells treated with PIPER, the mean TRF lengths decreased from 8.7 kb on day 6, to 7.7 kb on day 36, and to 6.5 kb on day 90 at the dose of 1.0 µM PIPER, while the mean TRF lengths decreased from 8.7 kb on day 6, to 7.7 kb on day 36, and to 6.4 kb on day 90 at the dose of 2.0 µM PIPER.
From the results above, the decrease in telomere length caused by both PM2 and PIPER in PC3 cells was much greater than that in LNCaP cells, probably because telomere lengths in PC3 cells are long (mean TRF length of 8.7 kb), while those in LNCaP cells are shorter (mean TRF length of 2.6 kb, for which some telomeres were already close to the critically short stage). With telomerase being inhibited, the long telomeres in the PC3 cells allowed most cells to proliferate, and telomere lengths were shortened accordingly. In contrast, more LNCaP cells were prone to become senescent in each cell passage, but the cells that proliferated were from cells with longer telomeres; therefore, the mean TRF length did not change much from the previous passages.
Another point of discussion would be why there were senescent cells in the PC3 culture, of which the average telomere length was much longer than that found in LNCaP cells. This observation can probably be explained by the heterogeneity of telomere lengths in PC3 cells, of which the TRFs can be seen spreading from over 21.2 to 4.2 kb, as shown in Fig. 4 and Supplementary Material Fig. S8. It was found that variable telomere lengths in prostate cancer cells and telomere shortening in cancer-associated stromal cells correlates with lethal disease.36) Since the shortest telomere, not the average telomere length, is critical for cell viability and the onset of replicative senescence,37,38) telomerase inhibition in PC3 cells could possibly inhibit the elongation of these few short telomeres and trigger these cells toward cellular senescence.
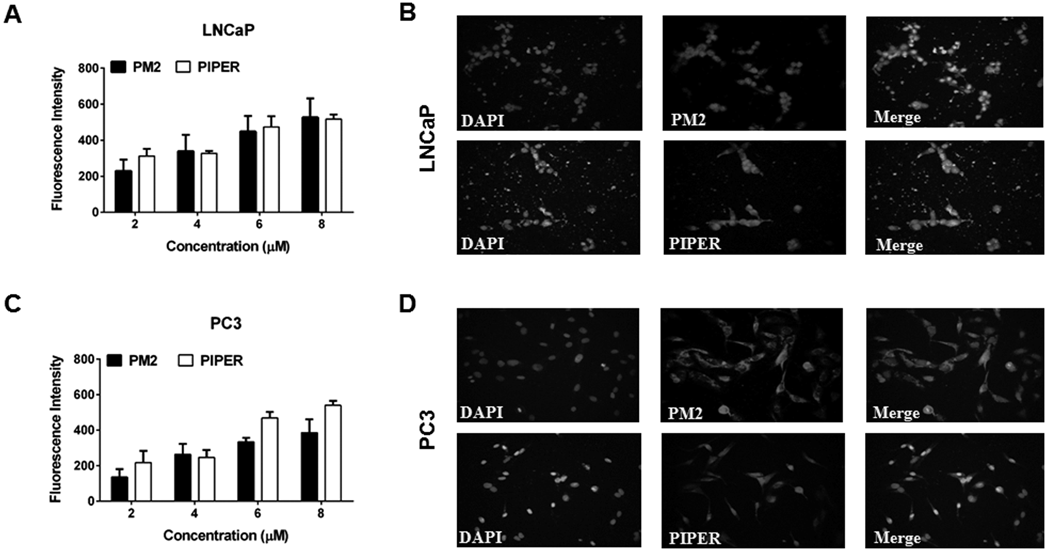
Partition Coefficient and Cellular Uptake of PM2 and PIPER in Prostate Cancer CellsFrom the experiments above, PM2 and PIPER induces telomere shortening and cellular senescence in both LNCaP and PC3 cells at a comparable concentration. However, the acute cytotoxicity test by SRB assay showed that PM2 is about 30 times more toxic than PIPER. We wondered whether cellular uptake of these two compounds might be different. First, we determined the octanol/H2O partition coefficient (Pow) of both compounds by the shake flask method. The Pow of PM2 and PIPER were found to be 1.5 ± 0.2 and 2.6 ± 0.1, respectively. Both compounds are lipophilic, but the Pow of PIPER is about one order of magnitude higher than that of PM2. We then measured the cellular uptake of PM2 and PIPER in LNCaP and PC3 cells using flow cytometry. As shown in Fig. 5A, both PM2 and PIPER appear to be absorbed into both LNCaP and PC3 cells in a concentration-dependent manner within the 24 h incubation time. Figure 5B shows pictures from fluorescence microscopy, of which LNCaP and PC3 cells were incubated with 4 µM of either PM2 or PIPER for 24 h before the cell nuclei were stained with DAPI. Both PM2 and PIPER appear to enter LNCaP and PC3 cells and distribute throughout the cells including the nucleus, where the fluorescent light merged with that of DAPI. The data from both flow cytometry and fluorescence microscopy suggest that both compounds are absorbed into the LNCaP and PC3 cells to a comparable extent, considering that they both have the same perylene chromophore. Therefore, the higher degree of cytotoxicity of PM2 over PIPER likely arises from mechanisms other than the cellular uptake of these compounds.
DISCUSSION
Prostate cancer is a slow-growing cancer, with a 5-year relative survival rate of 99%.2) Active surveillance for low-risk cancer has been increasingly adopted in the United States and internationally, and it is supported by various guidelines.39,40) Since most prostate cancers reactivate telomerase to circumvent the replicative senescence caused by the end replication problem, telomerase inhibition could be useful for the suppression of prostate cancer during this active surveillance. For a detectable solid tumor, a therapy regimen that combines telomerase inhibitors after conventional therapies such as surgery, radiotherapy, and chemotherapy, was suggested to be effective and durable responses for prostate cancer therapy.41) Sustained telomerase inhibition would lead the small population of dormant cancer cells, including cancer stem cells, to critical telomere attrition and ultimately cell senescence or cell death, with minimal impact on other normal somatic cells.41) However, this approach would be successful only if the telomerase inhibitor is safe to use for an extended period of time.
In the present study, we investigated two perylene derivatives, PM2 and PIPER, as effective telomerase inhibitors in two types of prostate cancer cells, LNCaP and PC3 cells. Both compounds were previously found to be G-quadruplex ligands that facilitate G-quadruplex formation of both telomeric DNA and the promoters of hTERT, leading to the inhibition of telomerase in an in vitro TRAP assay, and the suppression of hTERT gene expression and telomerase activity in A549 lung cancer cells.27) To investigate the effects of PM2 and PIPER during the early and late stages of prostate cancer, we chose LNCaP and PC3 prostate cancer cell lines to represent these stages. LNCaP cells represent an early stage of prostate cancer based on their source of origin (lymph node metastatic lesion of human prostate adenocarcinoma), androgen-sensitive cell growth, and the progression to AR-independent growth.42) PC3 cells represent an advanced stage of prostate cancer based on their source of origin (prostatic adenocarcinoma metastatic to bone), androgen-insensitive characteristics, and a more aggressive phenotype that is highly angiogenic.42) Our results showed that both PM2 and PIPER suppressed hTERT expression and telomerase activity in both types of cells. These compounds also suppressed gene expression of cancer-promoting genes such as c-Myc, VEGF, and AR, likely through the obstruction of the transcription machinery via G-quadruplex formation at their promoters. Long-term treatment with subcytotoxic doses of these two compounds led to telomere shortening and cellular senescence in both types of cells. However, the telomere shortening in LNCaP was not as noticeable as it was in PC3 due to the much shorter telomeres in LNCaP, which are probably close to the critical stage of triggering cellular senescence. Cellular senescence increased significantly in both cells during long-term treatment with either PM2 or PIPER, correlating well with the telomere shortening found in these cells. It is worth mentioning that in the controls set of both LNCaP and PC3 cells, there was a small increase in cellular senescence upon passages, which might reflect the insufficient telomerase-based elongation of telomeres or other senescence stimuli presence during cell passages. The short telomeres in LNCaP cells might be responsible for the genomic instability often observed in these cells, which leads to their androgen-independent progression upon passages.43)
An ideal telomerase inhibitor should exert its activity on telomerase with minimal effect on cell viability, which reflects certain mechanisms other than telomerase inhibition. PM2 is an asymmetrical perylene monoimide, while PIPER is a symmetrical perylene diimide. In the acute cytotoxicity test, PM2 was about 30 times more toxic than PIPER in both LNCaP and PC3 prostate cancer cells. In short-term treatments, PM2 appeared to be more effective than PIPER in the suppression of hTERT expression and telomerase activity in both types of prostate cancer cells. These results are similar to the results we previously found in A549 lung cancer cells.22) In long-term treatments, both PM2 and PIPER induced telomere shortening and cellular senescence at doses much closer in concentration to each other (0.4–0.8 µM of PM2 versus 1.0–2.0 µM of PIPER). The differences between PM2 and PIPER in the acute cytotoxicity and the short-term results from hTERT expression assay and telomerase activity assay prompted us to suspect whether these discrepancies were due to differences in the cellular uptake of these two compounds. PM2 is more water-soluble than PIPER; consequently, there might be more free molecules of PM2 available in solution at equivalent concentrations. PIPER is also well-known to aggregate in aqueous solution, especially at basic pH.23) In this study, we found that the partition coefficient (Pow) of PM2 and PIPER were 1.5 ± 0.2 and 2.6 ± 0.1, respectively. Therefore, both compounds are lipophilic, but the Pow for PIPER was about an order of magnitude higher than that for PM2. However, the difference in lipophilicity does not seem to affect the cellular uptake of both compounds into both LNCaP and PC3 cells. Using flow cytometry, we found that the uptake of both compounds into both types cancer cells occurred with similar intensity in a concentration-dependent manner, suggesting a passive absorption through the lipid bilayer of the cells. Fluorescence microscopy revealed that both compounds distributed throughout the whole cells, including in the nucleus. Although these two methods could not directly quantify the cellular uptake of the compounds, at least they imply a similar uptake of both compounds into both types of prostate cells in a similar manner (i.e., not with a 30-fold difference in cellular uptake). Therefore, the more acute cytotoxicity of PM2 is likely due to other cellular mechanisms, and PIPER might have a wider window of safety than PM2 when used as a telomerase inhibitor. It is also worth mentioning that both compounds were found to have little effect on peripheral blood mononuclear cells in the presence of 0–160 µM of either PM2 or PIPER.
When taken as a whole, these preliminary results in cell culture are encouraging, but it would be premature to extrapolate these results into clinical settings where many more factors need to be considered. The future animal and clinical trials will determine whether either of these compounds can safely prevent prostate cancer progression.
Acknowledgments
This work was supported by Grants from: (a) the Thailand Research Fund (RSA5880007), (b) the Royal Golden Jubilee Ph.D. (RGJ-PHD) Program (Grant No. PHD/0198/2556), (c) Faculty of Medicine Research Fund, Chiang Mai University, Chiang Mai, Thailand, (d) Graduate Student Supportive Fund, Faculty of Medicine, Chiang Mai University of the budget year 2014–2016 for Navakoon Kaewtunjai, and (e) the Robert A. Welch Foundation (Grant No. E-1320).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J. Clin., 65, 87–108 (2015).
- 2) Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J. Clin., 68, 7–30 (2018).
- 3) Zhang K, Bangma CH, Roobol MJ. Prostate cancer screening in Europe and Asia. Asian J. Urol., 4, 86–95 (2017).
- 4) Graham MK, Meeker A. Telomeres and telomerase in prostate cancer development and therapy. Nat. Rev. Urol., 14, 607–619 (2017).
- 5) Lee DJ, Mallin K, Graves AJ, Chang SS, Penson DF, Resnick MJ, Barocas DA. Recent changes in prostate cancer screening practices and epidemiology. J. Urol., 198, 1230–1240 (2017).
- 6) Fakhrejahani F, Madan RA, Dahut WL. Management options for biochemically recurrent prostate cancer. Curr. Treat. Options Oncol., 18, 26 (2017).
- 7) Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: a review. JAMA, 317, 2532–2542 (2017).
- 8) Cooperberg MR, Brooks JD, Faino AV, Newcomb LF, Kearns JT, Carroll PR, Dash A, Etzioni R, Fabrizio MD, Gleave ME, Morgan TM, Nelson PS, Thompson IM, Wagner AA, Lin DW, Zheng Y. Refined analysis of prostate-specific antigen kinetics to predict prostate cancer active surveillance outcomes. Eur. Urol., 74, 211–217 (2018).
- 9) Saad F, Fizazi K. Androgen deprivation therapy and secondary hormone therapy in the management of hormone-sensitive and castration-resistant prostate cancer. Urology, 86, 852–861 (2015).
- 10) Aragon-Ching JB, Dahut WL. Chemotherapy in androgen-independent prostate cancer (AIPC): what’s next after taxane progression? Cancer Ther., 5A, 151–160 (2007).
- 11) Marian CO, Wright WE, Shay JW. The effects of telomerase inhibition on prostate tumor-initiating cells. Int. J. Cancer, 127, 321–331 (2010).
- 12) Shay JW, Wright WE. Role of telomeres and telomerase in cancer. Semin. Cancer Biol., 21, 349–353 (2011).
- 13) Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol., 1, 72–76 (2000).
- 14) Victorelli S, Passos JF. Telomeres and cell senescence—Size matters not. EBioMedicine, 21, 14–20 (2017).
- 15) Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science, 266, 2011–2015 (1994).
- 16) Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR. Protein composition of catalytically active human telomerase from immortal cells. Science, 315, 1850–1853 (2007).
- 17) Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol. Mol. Biol. Rev., 66, 407–425 (2002).
- 18) Ramlee MK, Wang J, Toh WX, Li S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes, 7, 50 (2016).
- 19) Neidle S. Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J., 277, 1118–1125 (2010).
- 20) Palumbo SL, Ebbinghaus SW, Hurley LH. Formation of a unique end-to-end stacked pair of G-quadruplexes in the hTERT core promoter with implications for inhibition of telomerase by G-quadruplex-interactive ligands. J. Am. Chem. Soc., 131, 10878–10891 (2009).
- 21) Francisco AP, Paulo A. Oncogene expression modulation in cancer cell lines by DNA G-quadruplex-interactive small molecules. Curr. Med. Chem., 24, 4873–4904 (2017).
- 22) Mitchell T, Ramos-Montoya A, Di Antonio M, Murat P, Ohnmacht S, Micco M, Jurmeister S, Fryer L, Balasubramanian S, Neidle S, Neal DE. Downregulation of androgen receptor transcription by promoter G-quadruplex stabilization as a potential alternative treatment for castrate-resistant prostate cancer. Biochemistry, 52, 1429–1436 (2013).
- 23) Tassinari M, Cimino-Reale G, Nadai M, Doria F, Butovskaya E, Recagni M, Freccero M, Zaffaroni N, Richter SN, Folini M. Down-regulation of the androgen receptor by G-quadruplex ligands sensitizes castration-resistant prostate cancer cells to enzalutamide. J. Med. Chem., 61, 8625–8638 (2018).
- 24) Michelia E, D’Ambrosiob D, Franceschina M, Savino M. Water soluble cationic perylene derivatives as possible telomerase inhibitors: the search for selective G-quadruplex targeting. Mini Rev. Med. Chem., 9, 1622–1632 (2009).
- 25) D’Ambrosio D, Reichenbach P, Micheli E, Alvino A, Franceschin M, Savino M, Lingner J. Specific binding of telomeric G-quadruplexes by hydrosoluble perylene derivatives inhibits repeat addition processivity of human telomerase. Biochimie, 94, 854–863 (2012).
- 26) Fedoroff OY, Salazar M, Han H, Chemeris VV, Kerwin SM, Hurley LH. NMR-Based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry, 37, 12367–12374 (1998).
- 27) Taka T, Huang L, Wongnoppavich A, Tam-Chang SW, Lee TR, Tuntiwechapikul W. Telomere shortening and cell senescence induced by perylene derivatives in A549 human lung cancer cells. Bioorg. Med. Chem., 21, 883–890 (2013).
- 28) Tuntiwechapikul W, Taka T, Bethencourt M, Makonkawkeyoon L, Lee TR. The influence of pH on the G-quadruplex binding selectivity of perylene derivatives. Bioorg. Med. Chem. Lett., 16, 4120–4126 (2006).
- 29) Huang L, Tam-Chang SW. 9-Piperazine substituted perylene-3,4-dicarboximide as a fluorescent probe in ratiometric analysis. Chem. Commun., 47, 2291–2293 (2011).
- 30) Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc., 1, 1112–1116 (2006).
- 31) Szatmari I, Aradi J. Telomeric repeat amplification, without shortening or lengthening of the telomerase products: a method to analyze the processivity of telomerase enzyme. Nucleic Acids Res., 29, e3 (2001).
- 32) OECD guideline for the testing of chemicals. No. 107: Partition coefficient (n-octanol/water): shake flask method, OECD Publishing, Paris (1995).
- 33) Wattanasin P, Saetear P, Wilairat P, Nacapricha D, Teerasong S. Zone fluidics for measurement of octanol–water partition coefficient of drugs. Anal. Chim. Acta, 860, 1–7 (2015).
- 34) Taka T, Joonlasak K, Huang L, Randall Lee T, Chang SW, Tuntiwechapikul W. Down-regulation of the human VEGF gene expression by perylene monoimide derivatives. Bioorg. Med. Chem. Lett., 22, 518–522 (2012).
- 35) Sommerfeld HJ, Meeker AK, Piatyszek MA, Bova GS, Shay JW, Coffey DS. Telomerase activity: a prevalent marker of malignant human prostate tissue. Cancer Res., 56, 218–222 (1996).
- 36) Graham MK, Meeker A. Telomeres and telomerase in prostate cancer development and therapy. Nat. Rev. Urol., 14, 607–619 (2017).
- 37) Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell, 107, 67–77 (2001).
- 38) Xu Z, Duc KD, Holcman D, Teixeira MT. The length of the shortest telomere as the major determinant of the onset of replicative senescence. Genetics, 194, 847–857 (2013).
- 39) Chen RC, Rumble RB, Loblaw DA, Finelli A, Ehdaie B, Cooperberg MR, Morgan SC, Tyldesley S, Haluschak JJ, Tan W, Justman S, Jain S. Active surveillance for the management of localized prostate cancer (Cancer Care Ontario Guideline): American society of clinical oncology clinical practice guideline endorsement. J. Clin. Oncol., 34, 2182–2190 (2016).
- 40) Loeb S. Active surveillance for prostate cancer. Rev. Urol., 20, 101–103 (2018).
- 41) Marian CO, Shay JW. Prostate tumor-initiating cells: a new target for telomerase inhibition therapy? Biochim. Biophys. Acta, 1792, 289–296 (2009).
- 42) Dozmorov MG, Hurst RE, Culkin DJ, Kropp BP, Frank MB, Osban J, Penning TM, Lin HK. Unique patterns of molecular profiling between human prostate cancer LNCaP and PC-3 cells. Prostate, 69, 1077–1090 (2009).
- 43) Langeler EG, van Uffelen CJ, Blankenstein MA, van Steenbrugge GJ, Mulder E. Effect of culture conditions on androgen sensitivity of the human prostatic cancer cell line LNCaP. Prostate, 23, 213–223 (1993).