INTRODUCTION
The WHO reported that malignant tumor is now the second cause of death in the world.1) Although numerous chemotherapies have been developed for the treatment of malignant tumors, most of the anticancer drugs cause problems such as serious side effects.2) Cancer treatments by gene silencing with small RNAs including microRNA and small interfering RNA (siRNA) have been receiving a great attention because of their specific therapeutic effects.3) These RNAs specifically cleave target mRNA in the cytoplasm in a sequence-dependent manner.4–6) However, naked RNAs are degraded easily by RNases in the blood and can not penetrate cellular membranes by themselves because of their high hydrophilicity and negatively charged property. To solve these problems, researchers have developed various vectors for RNA delivery.7–10) However, the development of safe and efficient delivery systems to achieve effective cancer therapy is still a challenging task.
Cationic liposomes (CL) have been developed as one of non-virus vectors for siRNA delivery. Especially, pH-responsive CL carrying siRNA are expected as a drug candidate because standard CL have some disadvantages such as cytotoxicity and low transfection efficiency due to siRNA degradation in the lysosomes. We previously reported that pH-responsive polycationic lipid in lipoplexes, which are DNA–liposome complexes, promotes the interaction of lipoplex with cellular membranes and endosomal escape of the lipoplex by the proton sponge effect.11) Based on these benefits, we have synthesized a series of polycationic lipids such as cetyl-polyethylenimine[1800], dicetyl phosphate (DCP)-spermidine, and DCP-spermine for preparation of pH-responsive polycationic liposomes (PCL).12,13) siRNA formulated in these PCL shows high gene-transfer activity without severe cytotoxicity in vitro.14) Recently, we synthesized a dicetyl phosphate-tetraethylenepentamine conjugate (DCP-TEPA) as a pH-responsive polycationic lipid and prepared DCP-TEPA-based liposomes (TL) for efficient siRNA delivery.15) In addition, TL were decorated with polyethylene glycol (PEG)-grafted Arg-Gly-Asp (RGD-TL) or Ala-Pro-Arg-Pro-Gly (APRPG-TL) for active targeting to tumors via systemic administration.15,16) It is known that the RGD peptide specifically binds to integrin αvβ3.17) The APRPG peptide, a peptide originally identified in our lab by in vivo bio-panning, binds to vascular endothelial growth factor receptor-1 (VEGFR-1).18) Intravenous injection of siRNA against mammalian rapamycin target (mTOR) complexed with APRPG-TL significantly inhibits the growth of pulmonary tumors in mice.19) Moreover, intravenous injection of APRPG-TL carrying miR-499 suppresses target gene expression and the growth of subcutaneous tumors in mice.20) On the other hand, for systemic administration of siRNA, TL needed to be improved because of the requirement of a large amount of additional helper lipids such as dioleoylphosphatidyl-ethanolamine (DOPE) to promote their cellular uptake and endosomal escape, thus resulting in the need for an increased dose of total lipids.
In the present study, we used a dioleylphosphate–diethylenetriamine conjugate (DOP-DETA, Fig. 1A),21) which is an unsaturated lipid derivative of polycation, for preparation of pH-responsive liposomes. Since DOP-DETA has both 2 unsaturated carbon chains for membrane fusion and a relatively shorter polycationic hydrophilic head for endosomal escape, DOP-DETA-based liposomes (DL) can induce significant gene silencing without the inclusion of DOPE.21) DL have been shown to be a promising carrier capable of inducing gene silencing with low doses of siRNA in vitro.21) Here, in this present study we designed a new formulation for systemic injection of siRNA by modifying DL with PEG (PEG-DL) and studied a key determinant to obtain in vivo effects of PEGylation of DL. Two types of PEG-lipid conjugates, distearoylphosphatidylethanolamine (DSPE)-PEG and distearoylglycerol (DSG)-PEG, were used for PEGylation of DL; and their effects on the biodistribution of PEG-DL and siRNA in mice were compared. In addition, the gene-silencing effects of PEGylated DL carrying siRNA (PEG-DL/siRNA) in tumors were examined in mice to evaluate the potential of PEG-DL for systemic siRNA delivery.
MATERIALS AND METHODS
MaterialsDipalmitoylphosphatidylcholine (DPPC), cholesterol, and DSPE-PEG6000 were kindly donated by Nippon Fine Chemical Co. (Hyogo, Japan). DSPE-PEG5000 and DSG-PEG5000 were purchased from NOF Co. (Tokyo, Japan). All siRNAs including cholesterol-conjugated and fluorescence-labeled siRNAs were purchased from Hokkaido System Science Co. (Hokkaido, Japan). The nucleotide sequences of siRNA against mouse polo-like kinase 1 (siPLK1) with a 2-nucleotide overhang (underline) were 5′-CCA AAG GAA UUC CGA GAA ATT-3′ (sense) and 5′- UUU CUC GGA AUU CCU UUG GTT-3′ (antisense); those of siRNA against green fluorescent protein (siGFP), 5′-GGC UAC GUC CAG GAG CGC ACC-3′ (sense) and 5′-UGC GCU CCU GGA CGU AGC CUU-3′ (antisense); those of siRNA against luciferase (siLuc) with a 2-nucleotide overhang (underline), 5′-GCU AUG GGC UGA AUA CAA ATT-3′ (sense) and 5′-UUU GUA UUC AGC CCA UAG CTT-3′ (antisense); and those of the control siRNA with a 2-nucleotide overhang (underline), 5′-AUC CGC GCG AUA GUA CGU AUU-3′ (sense) and 5′-UAC GUA CUA UCG CGC GGA UUU-3′ (antisense). Alexa Fluor 750-labeled siRNA (AF750-siRNA) was obtained from Japan Bio Services Co., Ltd. (Saitama, Japan). Cholesterol was conjugated with siRNA at the 3′-prime of the sense strand. Phenylmethylsulfonyl fluoride (PMSF), leupeptin, aprotinin, pepstatin A, saponin, Triton X-100, and albumin from bovine-serum (BSA) were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Sodium dodecyl sulfate (SDS), GelRed, sucrose, sodium phosphate, citrate, ammonium acetate, sodium chloride and paraformaldehyde were procured from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
Preparation of DL/siRNA ComplexesDOP-DETA, DPPC, and cholesterol (2 : 1 : 2 as a molar ratio) were dissolved in tert-butyl alcohol and freeze–dried. DL were produced by the hydration of the freeze–dried lipids with UltraPure™ DNase/RNase-Free Distilled Water (RNase-free water; Invitrogen, Carlsbad, CA, U.S.A.). Sizing was performed by extrusion for 10 times through a polycarbonate membrane filter having 100-nm pores (Nucleopore, Maidstone, U.K.). The DL and siRNA were mixed and incubated for 20 min at room temperature to form DL/siRNA complexes. The molar ratio of DL (total lipid amount) to siRNA was 7000/1. The PEG modification was performed by mixing DL/siRNA with DSG- or DSPE-PEG5000 at 40°C for 10 min. The particle size and ξ-potential of the complexes diluted with RNase-free water were measured with a Zetasizer Nano ZS (Malvern, Worcs, U.K.).
Transmission Electron Microscope (TEM) ImagingDL/siRNA (total lipid concentration; 5 mM) in a volume of 5 µL was placed on a grid (Nisshin EM, Tokyo, Japan) and dehydrated with warm air. After this step was performed for 3 cycles, each sample was negatively stained with 10 µL of 1% ammonium molybdate for 1 min and air dried for 1 h. Samples were imaged with an HT7700 TEM System (Hitachi High-Technologies, Tokyo, Japan).
Cell CulturesHT1080 human fibrosarcoma cells constitutively expressing green fluorescent protein (HT1080-EGFP), previously established by ourselves,22) and colon26 NL-17 murine colon carcinoma cells, established and donated by Dr. Yamori,23) were cultured in D-MEM/Ham’s F-12 medium (Wako Pure Chemical Industries, Ltd.). Both cell lines were cultured in medium supplemented with 10% inactivated fetal bovine serum (FBS; AusGeneX, Oxenford, Australia), 100 units/mL penicillin, and 100 µg/mL streptomycin (MP Biomedicals) in a CO2 incubator.
Real-Time RT-PCRColon26 NL-17 cells (1.5 × 105 cells/3 mL/well on 6-well plate) were transfected with DL/siPLK1 (5 nM siRNA). Total RNA was extracted with TRIZOL LS Reagent (Invitrogen) according to the manufacturer’s instruction at 24 h after the transfection. The cDNA was generated from the total RNA samples (2 µg) by using a first-strand cDNA synthesis kit (GE Healthcare, Buckinghamshire, U.K.). In the presence of PLK1 (TaKaRa Bio, Shiga, Japan) or β-actin primers (TaKaRa Bio) and SYBR Premix Ex Taq II (TaKaRa Bio), real-time RT-PCR was performed with a Thermal Cycler Dice Real Time System (TaKaRa Bio). The nucleotide sequences of the primers of PLK1 were 5′-CTT CGC CCA AAT GCT TCG AGA T-3′ (forward) and 5′-TAG GCT GCG GTG AAT TGA AGA T-3′ (reverse); and those of β-actin, 5′-TGA CGG GGT CAC CCA CAC TGT GCC CAT CTA-3′ (forward) and 5′-CTA GAA GCA TTT GCG GTG GAC GAT GGA GGG-3′ (reverse). The conditions for real-time RT-PCR were as follow: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 60°C for 30 s, 95°C for 15 s, 60°C for 30 s, and 95°C for 15 s.
Western BlottingAnti-PLK1 rabbit polyclonal antibody (Cell Signaling Technology Japan, Tokyo, Japan) and anti-β-actin rabbit polyclonal antibody (Sigma-Aldrich) were diluted according to the manufacturer’s instructions. Colon26 NL-17 cells (1.5 × 105 cells/3 mL/well on 6-well plates) were transfected with DL/siPLK1. The culture medium was replaced with a fresh one at 24 h after the transfection. At 48 h after the transfection, the cells were lysed with lysis buffer (10 mM Tris–HCl buffer containing protease inhibitors and 0.1% SDS). Total protein content was measured with a BCA Protein Assay Reagent Kit (PIERCE Biotechnology, Rockford, IL, U.S.A.). The cell extracts were loaded into 10% SDS-polyacrylamide gel electrophoresis (PAGE) gels and separated by electrophoresis, and then transferred electrophoretically onto an Immobilon R-P Transfer Membrane (Merck Millipore, Billerica, MA, U.S.A.). After having been blocked for 1 h at room temperature with 5% BSA in Tris–HCl-buffered saline, pH 7.4, containing 0.1% Tween 20 (TTBS; Bio-Rad, Hercules, CA, U.S.A.), the membrane was incubated with a primary antibody (against PLK1 or β-actin) overnight at 4°C. Then, it was incubated with an horseradish peroxidase (HRP)-conjugated secondary antibody (Abcam, Cambridge, MA, U.S.A.) according to the manufacturer’s instructions. After the membrane had been washed with TTBS, each sample was developed with a chemiluminescent substrate (GE Healthcare), and each protein was detected with a LAS-3000 mini imaging system (FUJIFILM, Tokyo, Japan).
Evaluation of Anti-proliferation Effect of DL/siPLK1 on Colon26 NL-17 CellsColon26 NL-17 cells (1.0 × 103 cells/200 µL/well in a 96-well plate) were transfected with DL/siPLK1 (n = 6). The culture medium was replaced to the fresh one at 24 h after the transfection. The cell growth was evaluated at 0, 24, 48, 72, and 96 h after the transfection by using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) in accordance with the manufacturer’s instructions. The absorbance was measured by use of a Tecan Infinite M200 microplate reader (Tecan, Männedorf, Switzerland) operated at a test wavelength of 450 nm and a reference wavelength of 630 nm.
Physicochemical Characteristics of PEG-DL/siRNA in the Presence of SerumThe PEG modification was performed by mixing DL/siRNA with DSPE-PEG 6000 at 40°C for 10 min. The molar ratios, as a percentage, of PEGylation to total lipids were 2.5, 5 and 10%. Then, these samples were incubated in 50% FBS at 37°C for 1 h. After the incubation, the absorbance was measured by using the Tecan Infinite M200 microplate reader at 600 nm. Particle size of the complexes diluted with RNase-free water was measured by using a Zetasizer Nano ZS.
In Vivo Biodistribution of PEG-DL/siRNAWe added a trace amount of [3H]cholesteryl hexadecyl ether (GE Healthcare) to the initial tert-butyl alcohol solution for the preparation of radiolabeled DL.24,25) Five-week-old BALB/c male mice (n = 6) were injected with [3H]-labeled PEG-DL via a tail vein (74 kBq/mouse). Three or twenty-four hours after the injection, the mice were sacrificed under deep anesthesia with 2.0% isoflurane for collection of the blood. The post-heparin plasma was obtained by centrifugation (4°C, 700 × g, 10 min). Then, the heart, lungs, liver, spleen, and kidneys were removed, washed with saline, and weighed. The radioactivity in each organ and plasma was determined with a liquid scintillation counter (LSC-7400, Hitachi Aloka Medical, Tokyo, Japan). The total amount in the plasma was calculated based on the body weight of the mice, where the plasma volume was assumed to be 4.27% of the body weight based on the data for total blood volume.
Evaluation of the Stability of siRNA in the Presence of SerumNaked siRNA or siRNA formulated in DL or PEG-DL was incubated in 90% FBS at 37°C for 24 h. The siRNA was extracted from the serum by using TRIZOL LS Reagent and subjected to 15% polyacrylamide gel electrophoresis. Un-degraded siRNA was stained with GelRed, and the fluorescence was observed with an LAS-3000 mini imaging system under UV irradiation.
Electrophoretic AssaysiRNAs detached from DL during PEGylation were evaluated by an electrophoresis assay. siRNA or C-siRNA formulated in PEG-DL dissolved in Novex TBE-Urea sample buffer (Invitrogen) was applied to 15% polyacrylamide gel. After electrophoresis, free siRNA was stained with GelRed; and fluorescence was observed by using the LAS-3000 mini imaging system under UV irradiation.
Evaluation of RNA Interference (RNAi) Effect of C-siRNA by GFP Knockdown AssayHT1080-EGFP cells (2.0 × 104 cells/500 µL/well in a 24-well plate) were transfected with DL/C-siGFP. Twenty-four hours after the sample addition, the medium was exchanged for fresh medium; and the cells were further cultured for 24 h. The cells were then lysed with 1% n-octyl-β-D-glucoside containing protease inhibitors (1 mM PMSF, 2 µg/mL leupeptin, 2 µg/mL aprotinin, and 2 µM pepstatin A). After the cell lysis, the fluorescence intensities of GFP were measured by using the Tecan Infinite M200 microplate reader and corrected for protein amounts determined with a BCA Protein Assay Reagent Kit.
Experimental AnimalsFive-week-old BALB/c or C57BL/6 male mice were purchased from Japan SLC Inc. (Shizuoka, Japan). The animals were cared for according to the Animal Facility Guidelines of the University of Shizuoka. All animal experiments were approved by the Animal and Ethics Review Committee of the University of Shizuoka.
Ex Vivo Biodistribution of Fluorescence-Labeled siRNA after Administration with DL or PEG-DLColon26 NL-17 cells (1.0 × 106 cells/mouse) were implanted subcutaneously into the right flank of 5-week-old BALB/c male mice. The mice were fed an alfalfa-free feed (Oriental Yeast Co., Tokyo, Japan) to reduce the influence of background fluorescence. Ten days after the tumor implantation, the tumor-bearing mice were anesthetized continuously via inhalation of 2% isoflurane (Mylan Pharmaceuticals, Morgantown, WV, U.S.A.) and intravenously injected via a tail vein with AF750-siRNA or AF750-siRNA formulated in DL or PEG-DL (15 µg). The mice were subsequently sacrificed under anesthesia at 24 h post injection. Then, the organs (heart, lungs, liver, spleen, kidneys) and tumors were collected and imaged ex vivo with a Xenogen IVIS Lumina System coupled with Living Image software for data acquisition (Xenogen, Corp, Alameda, CA, U.S.A.).
Knockdown of Luciferase Expression in Vivo Using PEG-DL/C-siRNALL/2-luc-M38 cells (Bioware cell line, Caliper LifeScience, Waltham, MA, U.S.A., 1 × 106 cells/mouse) were implanted subcutaneously into the back of 5-week-old C57BL/6 male mice (n = 5 or 6). At day 9 after the tumor implantation (average tumor volume, 141.0 ± 42.5 mm3), the mice were given an intravenous (i.v.) injection of phosphate buffered saline (PBS) (control), PEG-DL/C-siCont or PEG-DL/C-siLuc (0.1 mg/kg as siRNA). For the assessment of luciferase gene expression, on day 0, 1 or 3 after the injection of each sample, luciferin (3 mg/mouse, Promega KK, Tokyo, Japan) was injected intraperitoneally (i.p.) and the luminescence intensity from the mice was measured 10 min post luciferin injection by the in vivo imaging system (IVIS). Tumor volume was measured by calipers and calculated by use of the following formula: a × b2 × 0.4 (a, largest diameter; b, smallest diameter).
StatisticsDifferences among groups were evaluated by ANOVA with the Tukey post-hoc test.
RESULTS AND DISCUSSION
Characteristics of DL/siRNAThe particle size and ξ-potential of DL and DL/siRNA were not significantly different (Table 1). The average size of these particles was approximately 150–160 nm, with a narrow size distribution (PdI = <0.100); and their ξ-potential was approximately 50 mV. Representative images obtained by transmission electron microscopy (TEM) showed that DL were round in shape and that their size distribution was uniform (Fig. 1B). DL carrying siRNA for PLK1, a serine/threonine protein kinase, induced the knockdown of its target mRNA and protein in colon26 NL-17 murine colon carcinoma cells in a sequence-dependent manner (Figs. 2A, B). Inhibition of the PLK1 protein expression was observed even at a low siRNA concentration (2.5 nM). Although TL, previously developed pH-responsive liposomes, require a large amount of DOPE to induce gene silencing efficiently,15) our newly developed DL induced gene silencing at low doses of siRNA even without DOPE. Because DOP-DETA contained unsaturated lipids in their structure, these liposomes might have facilitated membrane fusion between DL/siRNA and biomembranes. Our recent study also showed that DL induced a sufficient change in surface charge in response to a change in pH, resulting in efficient endosomal escape and gene silencing.21) DL/siPLK1 (1.0 nM siRNA) significantly suppressed the growth of colon26 NL-17 cells (Fig. 2C). Since PLK1 is a cell-cycle protein overexpressed in certain cancer cells,26–28) delivery of siPLK1 to cancer cells is a promising approach for cancer treatment.
Table 1. Characteristics of DL and DL/siRNA
| Size (d·nm) | PdI | ζ-Potential (mV) |
|---|
| DL | 152 ± 2 | 0.09 ± 0.04 | +50 ± 5 |
| DL/siRNA | 161 ± 12 | 0.07 ± 0.04 | +48 ± 5 |
Particle size and ζ-potential of liposomes diluted with RNase-free water were measured by use of a Zetasizer Nano ZS.
For the delivery of siRNA to a target tissue by intravenous injection, DL/siRNA was PEGylated to obtain stability in the blood and long-circulation property. In this study, 2 types of PEG-lipid conjugates, DSPE-PEG and DSG-PEG, were used to investigate the importance of the lipid structure of PEG-lipid derivatives. The structural difference between DSPE-PEG5000 and DSG-PEG5000 is shown in Fig. 3A. DSPE-PEG has a negatively charged phosphate group, while DSG-PEG does not. The biodistribution of PEG-DL/siRNA modified with DSPE-PEG or DSG-PEG was examined 3 h after intravenous injection into mice (Fig. 3B). The result showed that DL/siRNA modified with DSPE-PEG showed excellent blood retention compared with non-PEGylated DL/siRNA. However, DL/siRNA modified with DSG-PEG did not show such improvement of distribution. This result suggests that the electrostatic interaction between the phosphate group of DSPE-PEG and the amine group of DOP-DETA is important for PEG-lipid to remain stably on the surface of PEG-DL/siRNA. Since DL contained 40 mol% of DOP-DETA (unsaturated lipids), we considered that DSG-PEG was easily eliminated from the surface of DL in the blood. Thus, our most important conclusion is that the lipophilic interaction between DSG-PEG and lipids of nanoparticles was not sufficient for stable PEGylation to obtain a long-circulation property.
Based on these results, DSPE-PEG was used for PEGylation of DL/siRNA in subsequent experiments. Next, DL/siRNA were modified with 2.5, 5, or 10% DSPE-PEG6000. For this experiment, DSPE-PEG6000 was used for PEGylation instead of DSPE-PEG5000; because DL/siRNA modified with DSPE-PEG6000 showed longer circulation than that modified with DSPE-PEG5000 (data not shown). The particle size of PEG-DL/siRNA was not altered in the presence of serum, whereas that of non-PEGylated DL/siRNA was dramatically increased by it (Table 2). Similarly, although the turbidity of DL/siPLK1 (without PEGylation) was approximately 25-fold increased by incubation with 50% FBS, that of the PEG-DL/siPLK1 was not increased (Fig. 4A). These data indicate that PEGylation prevented aggregation of DL/siPLK1 in serum. In addition, the particle size of PEG-DL/siRNA seemed to be more suitable for long circulation and passive targeting to tumors via the enhanced permeability and retention (EPR) effect. The biodistribution of PEG-DL/siPLK1 at 24 h was evaluated after intravenous injection (Fig. 4B). The circulation of DL/siPLK1 was significantly improved by PEGylation in a PEG amount-dependent manner. About 20% of the injected PEG-DL/siRNA remained in the blood when DL/siRNA was modified with 10% DSPE-PEG. In addition, the accumulation of PEG-DL/siRNA in the liver and spleen was decreased by the 10% modification. In general, 10% PEG would seem to be a high amount for the modification of liposomes, but our previous study also indicated that a relatively high amount and long PEG chain length are effective to prolong the circulation time of lipoplexes.15) Masking of the surface property of lipoplexes with an adequate amount and length of PEG would be a key determinant to control their biodistribution. For these reasons, DL/siRNA modified with 10% DSPE-PEG was used for subsequent experiments.
Table 2. Size Distribution of Each PEG-DL
| FBS (−) | FBS (+) |
|---|
| Size (d·nm) | Size (d·nm) |
|---|
| PEG 0% | 146 ± 5 | 4750 ± 730 |
| PEG 2.5% | 156 ± 4 | 137 ± 0.8 |
| PEG 5% | 146 ± 1 | 122 ± 15 |
| PEG 10% | 96 ± 9 | 73 ± 0.7 |
PEG-DL/siRNA was incubated with 90% serum at 37°C for 24 h to test the protective effect of PEG-DL on siRNA degradation (Fig. 4C). Degradation of siRNA was prevented by use of PEG-DL/siRNA, whereas almost all naked siRNA and most of siRNA formulated in DL was degraded in the presence of serum. To explore siRNA biodistribution in mice bearing a subcutaneous tumor, we prepared AlexaFluor750-conjugated siRNA (AF750-siRNA) formulated in PEG-DL and intravenously injected them into mice. Fluorescence ex vivo imaging at 24 h post injection showed that AF750-siRNA formulated in PEG-DL accumulated in the colon26 NL-17 tumor to a greater extent compared with that in DL or naked siRNA (Fig. 4D). AF750-siRNA formulated in non-PEGylated DL accumulated in the liver and spleen more than that in PEG-DL, which is consistent with the data shown in Fig. 4B. These data suggest that systemically injected PEG-DL/siRNA could accumulate in solid tumors via the EPR effect.
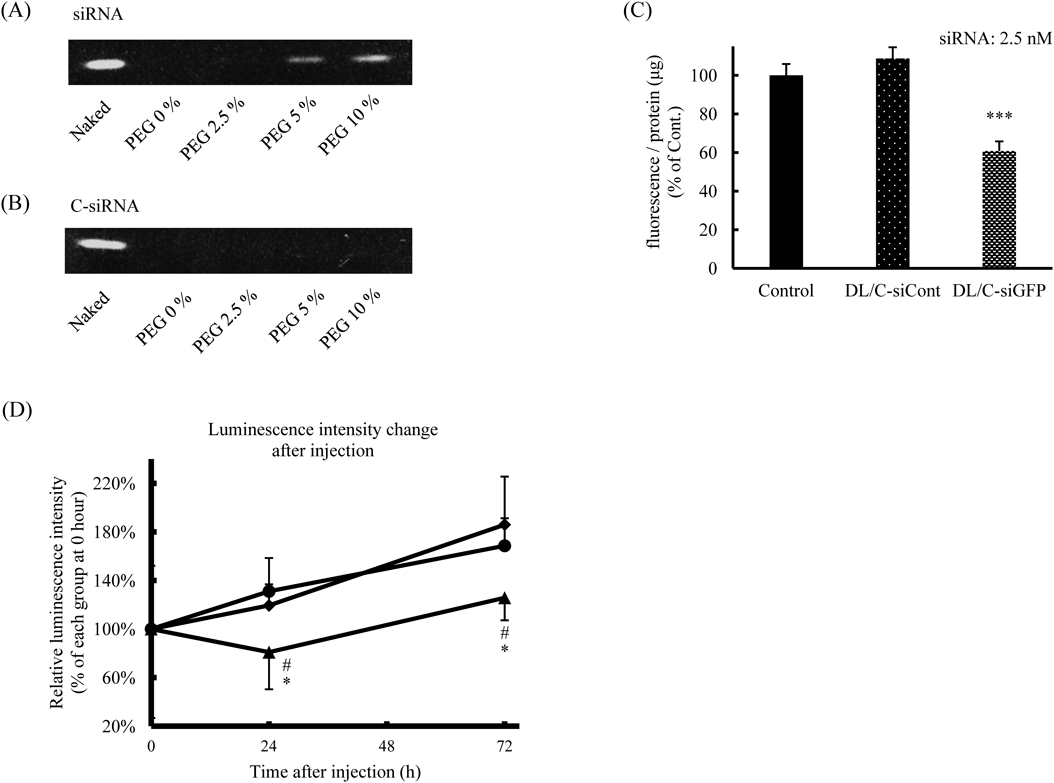
Protein Knockdown in Tumors after Systemic Injection of PEG-DL/siRNAWe previously reported that siRNA can be detached from liposomes during PEGylation.15) The phosphate group in DSPE-PEG may interfere with the electrostatic interaction between siRNA and DL. Therefore, the retention of siRNA in DL after PEGylation was examined by performing an electrophoretic assay. The results showed that a small amount of siRNA was detached from DL after modification with 10% DSPE-PEG (Fig. 5A). But we considered that most of the siRNA was not detached from PEG-DL and accumulated in the tumors (Fig. 4D). This detachment of siRNA from DL was prevented by conjugation of siRNA with cholesterol at the 3′-prime of the sense strand (C-siRNA), possibly through hydrophobic interaction between the cholesterol of C-siRNA and lipids of DL (Fig. 5B). The knockdown effect of C-siRNA formulated in DL was examined by performing a GFP silencing assay before the in vivo experiment. C-siRNA for GFP (siRNA: 2.5 nM) formulated in DL suppressed the expression of GFP in a sequence-dependent manner (Fig. 5C). The in vivo gene silencing effect of PEG-DL/C-siRNA was examined in mice bearing a subcutaneous tumor of LL/2-luc-M38 firefly luciferase-transduced mouse lung carcinoma. C-siRNA for luciferase (C-siLuc, 0.1 mg/kg) formulated in PEG-DL was intravenously injected into the tumor-bearing mice at 10 d after the tumor implantation. The luminescence intensities in the tumors were increased over the whole monitoring period in the control groups (Fig. 5D). In contrast, PEG-DL/C-siLuc treatment significantly suppressed the increase in luminescence intensity in the tumors. We hypothesized that DSPE-PEG was gradually detached from DL/siRNA in the body, which triggered the interaction between DL/siRNA and the tumor cells. In previous studies, the siRNA dosage was often set at around 1.0–10 mg/kg to achieve in vivo knockdown by systemic administration.29) However, in our present study significant knockdown of target protein was observed at only one-tenth to -hundredth siRNA concentration (0.1 mg/kg) by use of PEG-DL. Our recent study showed that DL efficiently interact with cell membrane and induce rapid endosomal escape, which result in potent gene silencing even at a low siRNA concentration.21) The efficient gene silencing in the tumors might be obtained by DL-mediated efficient delivery of siRNA to the cytoplasm. However, precise mechanism of gene silencing in vivo with PEG-DL/siRNA is unclear at the present. Our study suggests that PEG-DL could be a useful systemic siRNA vector to induce gene silencing in solid tumors.