Abstract
The pathophysiological mechanism of central post-stroke pain (CPSP) is complicated and not well understood. Recently, it has been reported that an increase in the levels of spinal nitric oxide synthetase (NOS) occurs in cerebral ischemia, and spinal NOS is involved in the development of neuropathic pain. The aim of this study was to elucidate the mechanism of spinal NOS signaling in the development of CPSP. Male ddY mice were subjected to 30-min long bilateral carotid artery occlusion (BCAO). The withdrawal responses to mechanical stimuli were significantly increased as determined with von Frey test on days 1 and 3 after BCAO. Protein expression of spinal N(G),N(G)-dimethylarginine dimethylaminohydralase 1 (DDAH1), a key enzyme involved in the metabolism of the endogenous NOS, increased on day 1 after BCAO, but not on day 3. Intrathecal (i.t.) injection of PD404182, a DDAH1 inhibitor, significantly suppressed mechanical allodynia on day 1, but not on day 3 after BCAO. In addition, i.t. administration of NG-nitro-L-arginine methyl ester (L-NAME), a non-selective NOS inhibitor, significantly blocked mechanical allodynia on days 1 and 3 after BCAO. Furthermore, BCAO-induced increment of spinal NOS activity was inhibited by the pretreatment with PD404182. These results suggest that mechanical allodynia in the early stage of CPSP is caused by increment of NOS activity through upregulated DDAH1 in the spinal cord.
INTRODUCTION
Cerebral stroke is a disease with poor prognosis and limited treatment options.1) Central post-stroke pain (CPSP) is a syndrome that develops secondary to cerebral stroke and clinically manifests as burning pain, hyperalgesia, or allodynia.2,3) It has been proposed that this syndrome develops due to central sensitization and hyperexcitability of nociceptive neurons in the central nervous system,2,4) however, the detailed mechanisms remain unclear. Furthermore, a beneficial treatment for CPSP has not been established yet.5–7) Therefore, it is of great importance to elucidate the onset mechanisms of CPSP and to establish new methods for the treatment of this condition.
It has been recently reported that cerebral infarction could elicit secondary degeneration in non-ischemic areas, such as the spinal cord.8–10) There is evidence that, in mice, nitric oxide synthetase (NOS) is upregulated in the spinal cord after stroke as follows. Generally, NOS is involved in the synthesis of L-citrulline and nitric oxide (NO) from L-arginine, and its activity is affected by various factors, such as ischemic stress or noxious stimuli. For example, it is shown that middle cerebral artery occlusion (MCAO) caused an increase in NOS activity in the ischemic area.11) MCAO induced blood-spinal cord barrier damage on both sides of the cervical spinal cord.12) In addition, bone cancer-induced mechanical allodynia upregulated NOS in the lumbar spinal cord.13) NOS knockout mice suppressed formalin- or nerve injury-induced pain.14,15) Our results also demonstrated that spinal NOS activity increased in the CPSP model in mice.9) The alternation of spinal NOS signaling may be involved in the exacerbation of ischemic stress damage and/or the development of chronic pain.
N(G),N(G)-Dimethylarginine dimethylaminohydrolase 1 (DDAH1) is a methylated arginine degrading enzyme which is present in various tissues of the central and peripheral regions.16) DDAH1 has recently been reported to metabolize asymmetric dimethylarginine (ADMA), an endogenous NOS inhibitor, suggesting that DDAH1 is involved in the modulation of NOS activity.17,18)
The goal of the present study was to investigate the involvement of DDAH1 in the activation of spinal NOS signaling in the early stage of the development of CPSP in mice.
MATERIALS AND METHODS
AnimalsAnimal studies were approved by the ethical review board of the Kobe Gakuin University (approval number: A17-09). Experiments were performed in ddY male mice (5 weeks old, 25–30 g) purchased from SLC (Shizuoka, Japan). The animals were housed under a 12-hour light/12-hour dark cycle with ad libitum access to food and water. This study was conducted in accordance with the basic principles of breeding and using laboratory animals adopted by the Japanese Pharmacological Society.
Central Post-stroke Pain ModelGeneration of transient global cerebral ischemia model mice was performed according to the method described above.19) Briefly, mice were deeply anesthetized with pentobarbital (60 mg/kg). The common carotid artery on both sides were obstructed for 30 min (bilateral carotid artery occlusion (BCAO)): by Sugita standard aneurysm clips (Mizuho Immunology Association, Tokyo, Japan). The same procedure without BCAO was performed in sham operation mice.
DrugsBCAO mice were administered PD404182 (a DDAH1 inhibitor; Tocris Bioscience, Inc., Bristol, U.K.) (10, 50, and 100 µg/mouse) or vehicle by intrathecal (i.t.) injection. PD404182 was dissolved in 100% dimethyl sulfoxide (DMSO; Cayman Chemical Company, Ann Arbor, MI, U.S.A.), and the solution was diluted with saline (0.1% DMSO final concentration) (vehicle). NG-nitro-L-arginine methyl ester (L-NAME), a nonselective NOS inhibitor, (Sigma-Aldrich, MO, U.S.A.) hydrochloride was dissolved in saline. Saline (10 µL) and L-NAME (100 and 300 µg/mouse) were treated through i.t. injection in mice. Hypodermic needle 1/6 (Natsume Seisakusho Co., Ltd., Tokyo, Japan) was intrathecally inserted between the L4 and L5 region, and advanced 6 mm into the lumbar enlargement of the spinal cord. In the experiments, we determined the concentration of the drugs mentioned above in reference to other reports.20,21)
von Frey TestMechanical allodynia in BCAO and sham mice was measured by the von Frey filament test as previously described, with some modifications.22) Before testing, mice were placed on a 5 × 5 mm wire mesh grid floor for 1 h. Each mouse was probed with 0.4 g von Frey filaments (Semmes–Weinstein von Frey Anesthesiometer, Tokyo, Japan) in the middle of the plantar surface of the hindpaw. Mechanical allodynia was defined by increments of withdrawal responses to tactile stimulation. On days 1 and 3 after BCAO, PD404182 and L-NAME were administered and von Frey filament test was performed at 10, 20, 30, and 60 min. The experimenter was not informed of the drug administered.
Western Blotting AnalysisWe performed Western blotting by previously reported.22) Briefly, spinal L4–L6 samples were homogenized in homogenization buffer (2 mM β-mercaptoethanol, 1 mM Na3VO4, 0.1 M Tris–HCl [pH 7.5], 2 M NaCl, 5 mM Benzamidine, 20 mM NaF, 5 mM imidazole, 1 mM p-nitrophenyl phosphate, Tween 20) and diluted with an equal volume of 2× sodium dodecyl sulfate (SDS) sample buffer (0.5 M Tris–HCl [pH 6.8], 10% SDS, 12% β-mercaptoethanol, 20% glycerol, 1% bromophenol blue). The sample was heated for 3 min at 97°C and protein samples (20 µg) were isolated through electrophoresis on 12% SDS-polyacrylamide gel and then moved onto nitrocellulose membranes (BioRad, Hercules, CA, U.S.A.) at 15 V for 50 min. Membranes were blocked (1 h at room temperature) in Tris-buffered saline (pH 7.6) with 0.1% Tween 20, and either 5% bovine serum albumin (BSA) (Sigma-Aldrich) for DDAH1 or 5% skim milk for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; loading control; Wako Pure Chemical Industries, Ltd., Osaka, Japan) was added. Membranes were incubated with each primary antibody (overnight at 4°C in their corresponding blocking solution). DDAH1 was detected by rabbit polyclonal primary antibodies (1 : 500; AbCam, Tokyo, Japan). GAPDH (1 : 20000) was detected by mouse monoclonal primary antibodies (Chemicon International Inc., Temecula, CA, U.S.A.). Blots were then incubated at 1 h room temperature with horseradish peroxidase-conjugated secondary antibody; anti-rabbit immunoglobulin G (IgG) (1 : 1000; KPL, Guildford, U.K.) as regards DDAH1, and anti-mouse IgG (1 : 10000; KPL) as regards GAPDH. Photochemical western immunoblotting substrate (Pierce; Thermo Scientific, Rockford, IL, U.S.A.) detected immunological bands. Light capture device (AE-6981; ATTO, Tokyo, Japan) visualized the bands. Behind normalization to the value for GAPDH, immunological bands was evaluated by Cs-Analyzer software (Ver. 3.0) (ATTO).
NOS AssayNOS activity of spinal L4–L6 samples was analyzed on days 1 and 3 after BCAO according to the method described above.22,23) Briefly, the spinal L4–L6 samples were homogenized in homogenization buffer (1 mM phenylmethanesulfonyl fluoride, 25.5 mM leupeptin, 1 µM pepstatin A, and 150 µM aprotinin was added to 1 mL of homogenize buffer [0.1 mM Tris–HCl (pH 7.5), 6.4 mL of Tween 20, 22.4 µL of β-mercaptoethanol, sterile water, 60 mg of p-nitrophenyl phosphate, 29.6 mg of Na3VO4, 125.2 mg of benzamide, 134.4 mg of NaF, and 54.4 mg of imidazole]). After homogenization in solution and centrifugation (15000 × g, 4°C, 15 min), the supernatant was collected. In turn, the supernatant of each sample was recovered and the protein concentration (mg/mL) was analyzed by a plate reader (absorption wavelength 450 nm, SH-1200, CORONA). Next, 200 µL of reaction buffer (50 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid, 0.5 mM ethylenediaminetetraacetic acid), 10 µL of reduced nicotinamide adenine dinucleotide phosphate (NADPH) Part A (NADP+, glucose 6-phosphate, L-arginine) and 10 µL of NADPH Part B (glucose 6-phosphate dehydrogenase) were added to each supernatant. The mixture was responsed at 37°C for 3 h. After 3 h, it was cooled with ice for 5 min and 10 µL of nitrate reductase was added. Briefly vortexed samples were incubated for 20 min, and centrifuged (15200 × g, 4°C, 5 min). 50 µL Color Reagent #1 (sulfanilamide in 3 N HCl) was added into nitrite standard and 100 µL of the obtained samples in 96-well plate. After the plate was vibrated for 5 min at room temperature, 50 µL Color Reagent #2 (N-(1-naphthyl) ethylenediamine dihydrochloride) was added. Next, the plate was vibrated for 5 min at room temperature, and analyzed by a plate reader (absorption wavelength 540 nm, SH-1200, CORONA, Ibaraki, Japan). The NOS activity of each sample is expressed in µM NOS activity/µg protein.
Statistical AnalysisData are presented as mean ± standard error of the mean (S.E.M.). Biochemical and behavioral data were analyzed by one-way ANOVA followed by Tukey’s test. The criterion for statistical significance was a p value of less than 0.05 and 0.01.
RESULTS
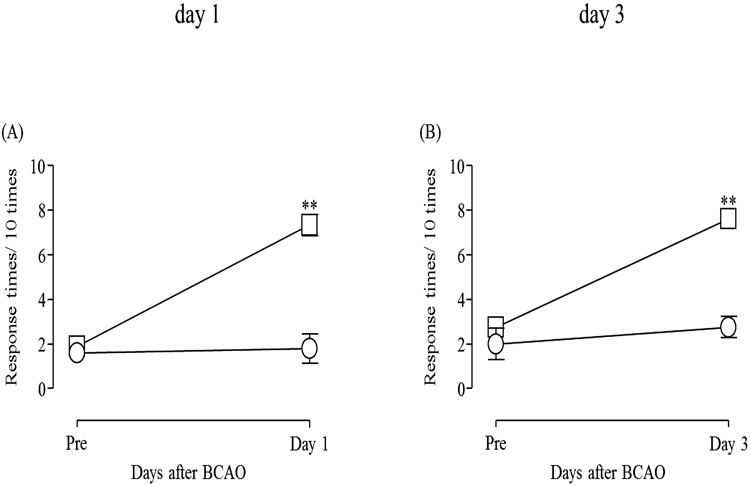
Development of Mechanical Allodynia on Days 1 and 3 after BCAOThe number of withdrawal responses significantly increased on days 1 and 3 after BCAO as compared with the sham control group (p < 0.01) (Fig. 1A: F(4, 8) = 1.100; Fig. 1B: F(3, 7) = 1.656)
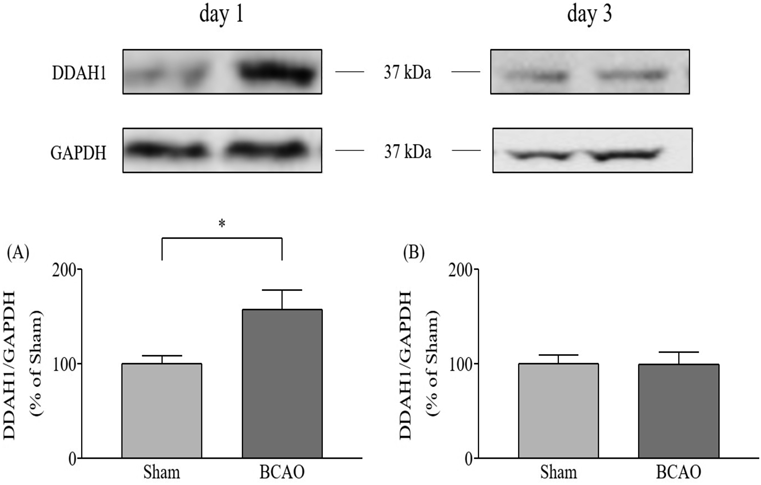
Expression of DDAH1 in the Spinal Cord after BCAOOn day 1 after BCAO, DDAH1 protein expression levels in the spinal cord were significantly increased compared with the sham control group (p < 0.05) (Fig. 2A: F(11, 11) = 5.891); however, this was not the case on day 3 after BCAO (Fig. 2B: F(14, 13) = 2.069).
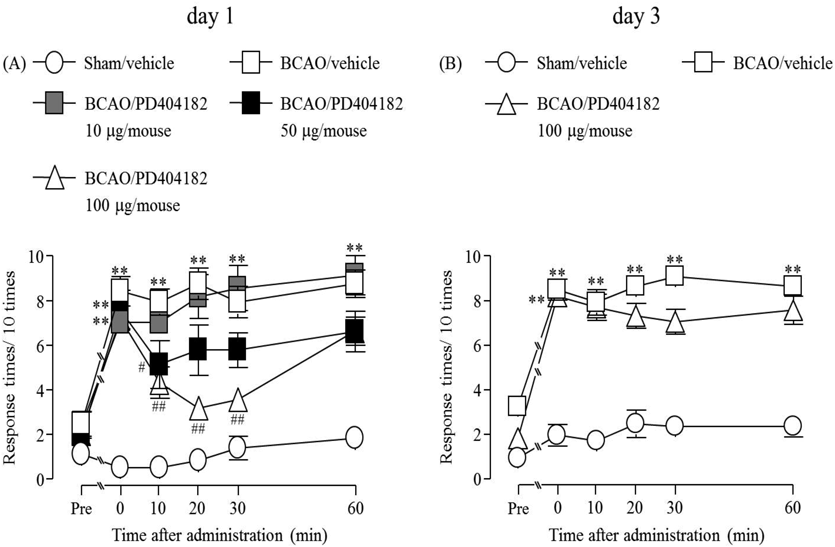
Effect of i.t. Administration of PD404182 on the Development of Mechanical Allodynia after BCAOA significant increase of the number of withdrawal responses determined by mechanical stimulation at times 0, 10, 20, 30 and 60 min was confirmed on days 1 and 3 after BCAO compared to the sham group (p < 0.01) (Fig. 3A, F(4, 25) = 9.8515; Fig. 3B, F(2, 15) = 15.398). This increase was dose-dependently suppressed by the pre-treatment with PD404182 (a DDAH1 inhibitor, 10, 50 and 100 µg/mouse) on day 1 after BCAO (p < 0.05, p < 0.01) (Fig. 3A). The suppressive effect of PD404182 did not persist at 60 min (Fig. 3A). However, the mechanical allodynia at 3 d after BCAO was not affected by the pre-treatment with PD404182 (100 µg/mouse) (Fig. 3B).
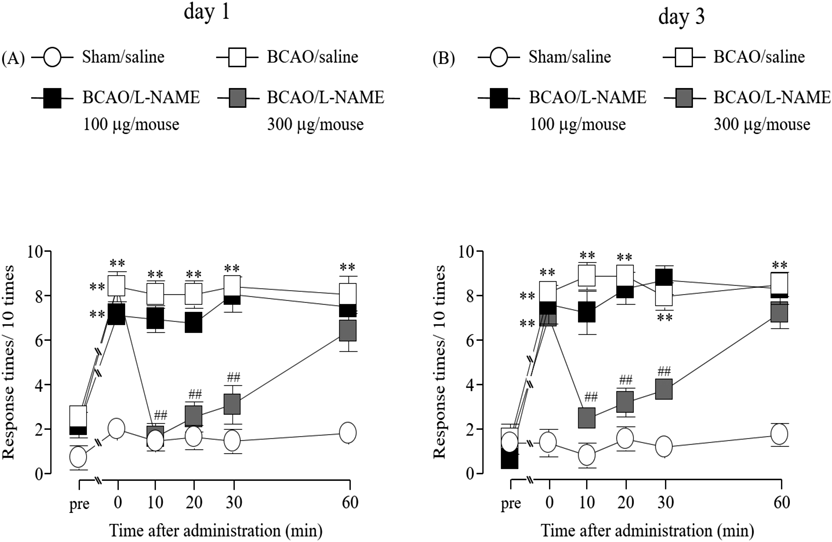
Effect of i.t. Administration of L-NAME on the Development of Mechanical Allodynia after BCAOOn days 1 and 3 after BCAO, withdrawal responses to mechanical stimuli were significantly suppressed by pre-administration of L-NAME (a nonselective NOS inhibitor, 300 µg/mouse) (p < 0.05, p < 0.01) within 30 min (Fig. 4A, F(3, 20) = 9.1789; Fig. 4B, F(3, 20) = 7.7103). On the other hand, L-NAME (100 µg/mouse) did not affect the mechanical stimuli (Figs. 4A, B).
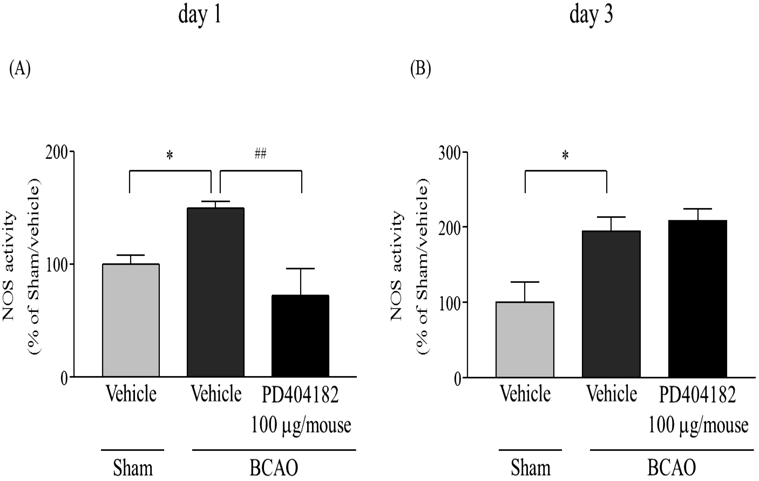
Effect of i.t. Administration of PD404182 on Increment of BCAO-Induced NOS Activity in the Spinal CordOn days 1 and 3 after BCAO, spinal NOS activity was significantly increased compared with the sham control group (p < 0.05) (Fig. 5A, F(2, 14) = 8.0679; Fig. 5B, F(2, 9) = 8.1971). Pre-treatment with PD404182 (100 µg/mouse) significantly suppressed spinal NOS activity on day 1 after BCAO (p < 0.01) (Fig. 5A). However, on day 3 after BCAO, spinal NOS activity was not affected by the PD404182 administration (Fig. 5B).
DISCUSSION
We found that spinal DDAH1 protein expression significantly increased on day 1 after BCAO. It is reported that DDAH1 is upregulated by inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β).24) Our previous study showed that activated spinal microglia is observed in BCAO model mice.9) In general, activated microglia releasing inflammatory cytokines is considered to be involved in the exacerbation of functional recovery after stroke and in the development of chronic pain.10,25,26) Therefore, it was possible that the upregulation of spinal DDAH1 may be induced by microglial activation, which was observed in BCAO model mice. Next, we found that the i.t. administration of PD404182 (a DDAH1 inhibitor) significantly ameliorates BCAO-induced mechanical allodynia on day 1. It has been reported that spinal administration of a DDAH1 inhibitor suppressed formalin-induced inflammatory pain.21) These results indicate that upregulation of spinal DDAH1 may be involved in the early phase of BCAO-induced mechanical allodynia.
On the other hand, on day 3 after BCAO, spinal DDAH1 protein expression did not change, indicating that BCAO-induced mechanical allodynia on day 3 might be affected by other pain related factors rather than by the mechanisms underlying DDAH1. Previously, it has been reported that high mobility group box-1 (HMGB1), which is a non-histone nuclear protein, regulated ischemia and neuropathic pain.27,28) The increment of HMGB1 induced the activation of NOS through nuclear factor-kappaB (NF-κB) signaling pathway activation in vitro.29) Possible mechanism is that HMGB1 signaling may cause the NOS upregulation in the late stage of CPSP. Because, our previous study have shown that increment of spinal HMGB1 signaling induced upregulation of NOS activity on day 3 after BCAO.9)
It has been previously reported that the i.t. administration of L-NAME completely reversed the reduction of paw withdrawal threshold induced by intraperitoneal administration of oxaliplatin.20) Sciatic nerve ligature led to a significant decrease of the threshold for evoked paw withdrawal to mechanical stimuli in wild-type mice; this response was abolished in mice lacking neuronal NOS (nNOS) or inducible NOS (iNOS).30) Similarly, we found that the i.t. administration of L-NAME markedly suppressed BCAO-induced mechanical allodynia. Therefore, one possible explanation is that the changes in NO levels and in NOS activity may be caused immediately after BCAO and may result in BCAO-induced mechanical allodynia.
We also demonstrated that NOS activity was increased in the spinal cord during pain induced by BCAO, and its elevation was significantly suppressed after i.t. administration of PD404182 on day 1 after BCAO. However, on day 3 after BCAO, i.t. administration of PD404182 did not suppress BCAO-induced spinal NOS activity upregulation. It is known that DDAH1 modulates NO signaling by catalyzing the hydrolysis of ADMA, an endogenous inhibitor of NOS,31) and the inhibition of DDAH1 significantly increased intracellular levels of ADMA and reduced NO production.32) It is reported that overexpression of DDAH1 elevated expression of nNOS and iNOS in vitro.33,34) DDAH1 knockdown decreased of iNOS expression in vitro.35) From these references, nNOS and iNOS signaling system is involved in the increment of NOS activity which induced by upregulation of DDAH1. Therefore, it is thought that the increase of spinal DDAH1 may be involved in the development of BCAO-induced mechanical allodynia through enhancement of NOS signaling.
In summary, DDAH1 was upregulated in the spinal cord in the early stage of BCAO model mice. The inhibition of DDAH1 led to suppression of BCAO-induced mechanical allodynia and spinal NOS activity in the early stage of the development of CPSP. Hence, the mechanical allodynia in the early stage of CPSP is likely caused by the increased NOS activity via upregulated DDAH1 expression in the spinal cord.
Acknowledgments
This work was supported by Grants-in-Aid and by special coordination funds from Grants-in-Aid for Scientific Research (C) [Grant number 16K10988] from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Henninger N, Kumar R, Fisher M. Acute ischemic stroke therapy. Expert Rev. Cardiovasc. Ther., 8, 1389–1398 (2010).
- 2) Kumar B, Kalita J, Kumar G, Misra UK. Central poststroke pain: a review of pathophysiology and treatment. Anesth. Analg., 108, 1645–1657 (2009).
- 3) Hong JH, Choi BY, Chang CH, Kim SH, Jung YJ, Lee DG, Kwon YH, Jang SH. The prevalence of central poststroke pain according to the integrity of the spino–thalamo–cortical pathway. Eur. Neurol., 67, 12–17 (2012).
- 4) Bowsher D, Leijon G, Thuomas KA. Central poststroke pain: correlation of MRI with clinical pain characteristics and sensory abnormalities. Neurology, 51, 1352–1358 (1998).
- 5) Lampl C, Yazdi K, Röper C. Amitriptyline in the prophylaxis of central poststroke pain. Preliminary results of 39 patients in a placebo-controlled, long-term study. Stroke, 33, 3030–3032 (2002).
- 6) Leijon G, Boivie J. Central post-stroke pain—a controlled trial of amitriptyline and carbamazepine. Pain, 36, 27–36 (1989).
- 7) Rasche D, Rinaldi PC, Young RF, Tronnier VM. Deep brain stimulation for the treatment of various chronic pain syndromes. Neurosurg. Focus, 21, E8 (2006).
- 8) Dang G, Chen X, Zhao Y, Chen Y, Ouyang F, Liang J, Guo Y, Zeng J. Alterations in the spinal cord and ventral root after cerebral infarction in non-human primates. Restor. Neurol. Neurosci., 36, 729–740 (2018).
- 9) Matsuura W, Harada S, Liu K, Nishibori M, Tokuyama S. Evidence of a role for spinal HMGB1 in ischemic stress-induced mechanical allodynia in mice. Brain Res., 1687, 1–10 (2018).
- 10) Dang G, Chen X, Chen Y, Zhao Y, Ouyang F, Zeng J. Dynamic secondary degeneration in the spinal cord and ventral root after a focal cerebral infarction among hypertensive rats. Sci. Rep., 6, 22655 (2016).
- 11) Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J. Cereb. Blood Flow Metab., 15, 378–384 (1995).
- 12) Garbuzova-Davis S, Haller E, Tajiri N, Thomson A, Barretta J, Williams SN, Haim ED, Qin H, Frisina-Deyo A, Abraham JV, Sanberg PR, Van Loveren H, Borlongan CV. Blood-spinal cord barrier alterations in subacute and chronic stages of a rat model of focal cerebral ischemia. J. Neuropathol. Exp. Neurol., 75, 673–688 (2016).
- 13) Yang Y, Zhang J, Liu Y, Zheng Y, Bo J, Zhou X, Wang J, Ma Z. Role of nitric oxide synthase in the development of bone cancer pain and effect of L-NMMA. Mol. Med. Rep., 13, 1220–1226 (2016).
- 14) Hervera A, Negrete R, Leánez S, Martín-Campos JM, Pol O. The spinal cord expression of neuronal and inducible nitric oxide synthases and their contribution in the maintenance of neuropathic pain in mice. PLoS ONE, 5, e14321 (2010).
- 15) Kuboyama K, Tsuda M, Tsutsui M, Toyohara Y, Tozaki-Saitoh H, Shimokawa H, Yanagihara N, Inoue K. Reduced spinal microglial activation and neuropathic pain after nerve injury in mice lacking all three nitric oxide synthases. Mol. Pain, 7, 50 (2011).
- 16) Zhou J, Xiong Q, Chen H, Yang C, Fan Y. Identification of the spinal expression profile of non-coding rnas involved in neuropathic pain following spared nerve injury by sequence analysis. Front. Mol. Neurosci., 10, 91 (2017).
- 17) Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, Vallance P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J., 343, 209–214 (1999).
- 18) Cooke JP, Ghebremariam YT. DDAH says NO to ADMA. Arterioscler. Thromb. Vasc. Biol., 31, 1462–1464 (2011).
- 19) Tamiya S, Yoshida Y, Harada S, Nakamoto K, Tokuyama S. Establishment of a central post-stroke pain model using global cerebral ischaemic mice. J. Pharm. Pharmacol., 65, 615–620 (2013).
- 20) Mihara Y, Egashira N, Sada H, Kawashiri T, Ushio S, Yano T, Ikesue H, Oishi R. Involvement of spinal NR2B-containing NMDA receptors in oxaliplatin-induced mechanical allodynia in rats. Mol. Pain, 7, 8 (2011).
- 21) DʼMello R, Sand CA, Pezet S, Leiper JM, Gaurilcikaite E, McMahon SB, Dickenson AH, Nandi M. Dimethylarginine dimethylaminohydrolase 1 is involved in spinal nociceptive plasticity. Pain, 156, 2052–2060 (2015).
- 22) Matsuura W, Harada S, Liu K, Nishibori M, Tokuyama S. Evidence of a role for spinal HMGB1 in ischemic stress-induced mechanical allodynia in mice. Brain Res., 1687, 1–10 (2018).
- 23) Nawa A, Fujita (Hamabe) W, Tokuyama S. Inducible nitric oxide synthase-mediated decrease of intestinal P-glycoprotein expression under streptozotocin-induced diabetic conditions. Life Sci., 86, 402–409 (2010).
- 24) Zhang P, Fassett JT, Zhu G, Li J, Hu X, Xu X, Chen Y, Bache RJ. Repetitive ischemia increases myocardial dimethylarginine dimethylaminohydrolase 1 expression. Vasc. Med., 22, 179–188 (2017).
- 25) Ji R-R, Xu Z-Z, Gao Y-J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov., 13, 533–548 (2014).
- 26) Qi J, Rong Y, Wang L, Xu J, Zhao K. Rab7b overexpression–ameliorated ischemic brain damage following tMCAO involves suppression of TLR4 and NF-κB p65. J. Mol. Neurosci., 68, 163–170 (2019).
- 27) Liu F, Wang Z, Qiu Y, Wei M, Li C, Xie Y, Shen L, Huang Y, Ma C. Suppression of MyD88-dependent signaling alleviates neuropathic pain induced by peripheral nerve injury in the rat. J. Neuroinflammation, 14, 70 (2017).
- 28) Ye Y, Zeng Z, Jin T, Zhang H, Xiong X, Gu L. The role of high mobility group box 1 in ischemic stroke. Front. Cell. Neurosci., 13, 127 (2019).
- 29) Das N, Dewan V, Grace PM, Gunn RJ, Tamura R, Tzarum N, Watkins LR, Wilson IA, Yin H. HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Reports, 17, 1128–1140 (2016).
- 30) Hervera A, Negrete R, Leánez S, Martín-Campos JM, Pol O. The spinal cord expression of neuronal and inducible nitric oxide synthases and their contribution in the maintenance of neuropathic pain in mice. PLoS ONE, 5, e14321 (2010).
- 31) Leiper JM. The DDAH-ADMA-NOS pathway. Ther. Drug Monit., 27, 744–746 (2005).
- 32) Ghebremariam YT, Erlanson DA, Cooke JP. A novel and potent inhibitor of dimethylarginine dimethylaminohydrolase: a modulator of cardiovascular nitric oxide. J. Pharmacol. Exp. Ther., 348, 69–76 (2013).
- 33) Luo Y, Yue W, Quan X, Wang Y, Zhao B, Lu Z. Asymmetric dimethylarginine exacerbates Aβ-induced toxicity and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic. Biol. Med., 79, 117–126 (2015).
- 34) Wang S, Hu C-P, Yuan Q, Zhang W-F, Zhou Z, Nie S-D, Jiang J-L, Li Y-J. Dimethylarginine dimethylaminohydrolase 1 regulates nerve growth factor-promoted differentiation of PC12 cells in a nitric oxide-dependent but asymmetric dimethylargenine-independent manner. J. Neurosci. Res., 90, 1209–1217 (2012).
- 35) Wang H, Guo Y, Liu L, Guan L, Wang T, Zhang L, Wang Y, Cao J, Ding W, Zhang F, Lu Z. DDAH1 plays dual roles in PM2.5 induced cell death in A549 cells. Biochim. Biophys. Acta, Gen. Subj., 1860, 2793–2801 (2016).