Regular Articles
Evaluation of Methods to Assess CYP3A Induction Risk in Clinical Practice Using in Vitro Induction Parameters
2021 年 44 巻 3 号 p. 338-349
詳細
2021 年 44 巻 3 号 p. 338-349
Established guidelines have recommended a number of methods based on in vitro data to assess the CYP3A induction risk of new chemical entities in clinical practice. In this study, we evaluated the predictability of various assessment methods. We collected in vitro parameters from a variety of literature that includes data on 19 batches of hepatocytes. Clinical CYP3A induction was predicted using 3 direct approaches—the fold-change, basic model, and mechanistic static models—as well as 5 correlation approaches, including the relative induction score (RIS) and the relative factor (RF) method. These predictions were then compared with data from 30 clinical inductions. Collected in vitro parameters varied greatly between hepatocyte batches. Direct assessment methods using fixed cut-off values provided a lot of false predictions due to hepatocyte variability, which can overlook induction risk or lead to needless clinical drug–drug interaction (DDI) studies. On the other hand, correlation methods with the cut-off values set for each batch of hepatocytes accurately predicted the induction risk. Among these, the AUCu/inducer concentrations for half the maximum induction (EC50) and the RF methods which use the area under the curve (AUC) of the unbound inducers for calculating induction potential showed an especially good correlation with clinical induction. Correlation methods were better at predicting clinical induction risk than the other methods, regardless of hepatocyte variability. The AUCu/EC50 and the RF methods in particular had a small number of false predictions, and can therefore be used to assess induction risk along with the other correlation methods recommended in guidelines.
Pharmaceutical industries are required to evaluate the potential for drug candidates to induce CYP enzymes because this can significantly decrease the exposure and efficacy of substrate drugs.1) Among these, CYP3A4 is a main metabolic enzyme for many commercial drugs in both the liver and the small intestine,2) so being able to predict clinical CYP3A induction ahead of first-in-human studies is especially important.
As recommended in the U.S. Food and Drug Administration (FDA) guidance, European Medicines Agency (EMA) guideline and the Ministry of Health, Labour and Welfare of Japan (MHLW) guideline, CYP induction risk should be assessed with in vitro experiments.3–5)
First, the fold increase in mRNA expression of CYP enzymes in the presence of test compounds at the expected in vivo concentration range is measured to identify the induction potential. If the increase is over 2-fold and more than 20% of the positive control, rifampicin, test compounds are regarded to have induction potential.
After that, by obtaining the maximum induction (Emax) and inducer concentrations for half the maximum induction (EC50) of the test compounds, several approaches can be used to more precisely predict clinical induction. The basic model is the simplest method; it directly compares the values calculated by using in vitro Emax, EC50, and clinical plasma concentrations of test compounds with the uniform threshold of drug–drug interaction (DDI) risk. Despite their simplicity, the fold increase in mRNA and predicted values given by the basic model method vary between experiments, which may result in false predictions. This is because the induction responses in primary cultured hepatocytes—the gold-standard of in vitro systems—show large inter-batch variability. Although induction data obtained using cell lines such as HepaRG6) or immortalized human hepatocytes (HepatoCells or Fa2N4)7,8) show minimal variability, they can only be used as supporting information. To avoid the false-negative prediction, total plasma concentrations are recommended for use in the basic model, but this may increase false-positives. Recently, the FDA and Pharmaceuticals and Medical Devices Agency (PMDA) revised the DDI guidance to use unbound plasma concentrations in order to decrease false-positive predictions. Because of the increase in false-negatives when directly using unbound plasma concentrations, a 10-fold scaling factor is recommended.9) In the present study, we examined and compared the old and revised calculation methods in the basic model.
Correlation approaches such as the Relative Induction Score (RIS),10) the maximum unbound plasma concentration of the inducers (Cmax,u)/EC50,1) and AUCu/EC5011) methods set thresholds for individual batches of hepatocytes by drawing a calibration curve with a set of known inducers. Setting individual thresholds might minimize inter-batch hepatocyte variability. Previous studies reported that correlation methods showed better correlations with clinical induction.12,13) However, it takes a lot of experiments to obtain calibration curves for each batch of hepatocytes. Furthermore, a standard set of known inducers for calibration is not specified, so prediction results might vary between laboratories according to the choice of inducers.
The mechanistic static and dynamic approaches are also options that can assess CYP enzyme-mediated metabolism in the liver and the intestine.14) Although they can predict the clinical induction considering the effect of physicochemical properties of the substrate drugs, they need various in vitro and in vivo inputs to do so.
Unfortunately, for compounds with poor solubility and/or cell toxicity, it is difficult to reach the expected worst case in vivo concentration range, including a 30- or 50-fold of Cmax,u3–5) and to calculate in vitro Emax and EC50 values. When in vitro risk assessment methods given in guidelines cannot be applied to a new drug candidate, the clinical induction study should be performed.
There are two reported correlation methods which do not need Emax and EC50. One is relative factor (RF) method15) which uses only the induction detection limit concentrations (IDLCs) of new chemical entities and standard inducers to evaluate CYP3A4 induction potential. The IDLC represents the compound concentration that induces the enzyme at 3-fold standard deviation (S.D.) from the control level. The other is the AUC/F2 method16) which uses only concentrations at which new chemical entities and standard inducers increase CYP3A4 mRNA level by 2-fold in HepaRG cells (F2). Both methods require experiments with standard inducers like the other correlation methods, but the number of experiments can be reduced because fewer points of induction are needed.
In this study, we collected in vitro induction data of known CYP3A inducers and non-inducers from a variety of literature for use in assessing the risk of CYP3A induction. We then compared the results of methods recommended by guidelines and also RF and AUC/F2 methods using a common in vitro induction dataset.
The Emax and EC50 values based on the CYP3A4 induction at the mRNA level were collected for 9 compounds, which are known to be clinical CYP3A inducers and non-inducers according to the decrease in AUC of co-administered substrate drugs in clinical studies.2,10,12,17–24) Compounds which also inhibit CYP3A at clinically relevant concentrations were excluded from this analysis to simply evaluate the induction effect. Total experimental data were from 19 batches of primary cultured human hepatocytes, 3 batches of HepaRG, 3 batches of HepatoCells, and 1 batch of Fa2N4.
In Vivo DataAs for inducers, the maximum total plasma concentrations (Cmax), the apparent oral clearance (CL/F) and the unbound fraction in plasma (fu) of inducers were collected from literature (Table 1). Cmax,u was calculated by using equation 1.
 | (1) |
| Inducer | CL/F (mL/min/kg) | fu | Cmax (µM) | Dose (mg) | Ref. |
|---|---|---|---|---|---|
| Rifampicin | 3.5 | 0.25 | 7.9 | 600 | 36) |
| Phenobarbital | 0.062 | 0.49 | 56 | 90 | 36) |
| Carbamazepine | 1.3 | 0.26 | 39 | 1288 | 36) |
| Phenytoin | 0.069 | 24 | 200 | 37) | |
| Rifapentine | 0.023 | 2.9 | 150 | 38,39) | |
| 0.023 | 7.8 | 300 | |||
| 0.023 | 20 | 600 | |||
| Pioglitazone | 1.2 | 0.01 | 4.5 | 45 | 36) |
| Nifedipine | 14 | 0.04 | 0.23 | 10 | 36) |
| Omeprazole | 14 | 0.05 | 0.68 | 20 | 36) |
| Rosiglitazone | 0.68 | 0.002 | 1.7 | 8 | 36) |
The area under the curve (AUC) of the total and unbound inducers was calculated by using equations 2 and 3, respectively.
 | (2) |
 | (3) |
The average steady-state unbound plasma concentration of inducers (Css,u) was calculated by the following equation, where τ is the dosage interval:
 | (4) |
As for substrates, the fraction of systemic clearance of the substrate mediated by CYP3A (fm) and intestinal bioavailability (Fg) were collected from literature21,25–27) (Supplementary Table 1).
We also collected data on plasma AUC decrease of CYP3A substrates from 30 clinical induction studies (23 studies for midazolam, 3 studies for ethinyl estradiol, 1 study for alprazolam, 2 studies for nifedipine and 1 study for simvastatin; Table 2). We defined positive CYP induction as when AUC decrease was more than 20% as designated by the FDA, EMA, and MHLW.3–5)
| Inducer | Dosing regimen | % of AUC decrease | Substrate | Ref. |
|---|---|---|---|---|
| Rifampicin | 10 mg q.d., 14 d | 50 | Midazolam | 17) |
| 20 mg q.d., 14 d | 62 | Midazolam | ||
| 100 mg q.d., 14 d | 75 | Midazolam | ||
| 600 mg q.d., 14 d | 51 | Midazolam | 18) | |
| 600 mg q.d., 14 d | 92 | Midazolam | ||
| 600 mg q.d., 5 d | 48 | Midazolam | 2) | |
| 600 mg q.d., 5 d | 95 | Midazolam | ||
| 5 mg q.d., 5 d | 17 | Midazolam | 19) | |
| 10 mg q.d., 5 d | 25 | Midazolam | ||
| 25 mg q.d., 5 d | 38 | Midazolam | ||
| 75 mg q.d., 5 d | 41 | Midazolam | ||
| 5 mg q.d., 5 d | 23 | Midazolam | ||
| 10 mg q.d., 5 d | 35 | Midazolam | ||
| 25 mg q.d., 5 d | 62 | Midazolam | ||
| 75 mg q.d., 5 d | 77 | Midazolam | ||
| Phenobarbital | 75 mg | 70 | Ethinyl estradiol | 10) |
| Carbamazepine | 600 mg q.d., 21 d | 94 | Midazolam | 12) |
| 600 mg q.d., 21 d | 42 | Ethinyl estradiol | 21) | |
| 100 mg t.i.d., 10 d | 58 | Alprazolam | 20) | |
| Phenytoin | 200 mg q.d., 17 d | 94 | Midazolam | 12) |
| Rifapentine | 450 mg q.d., 14 d | 91 | Midazolam | 22) |
| 750 mg q.d., 14 d | 91 | Midazolam | ||
| 1200 mg q.d., 14 d | 93 | Midazolam | ||
| 1650 mg q.d., 14 d | 93 | Midazolam | ||
| Pioglitazone | 45 mg q.d., 24 d | 26 | Midazolam | 23) |
| Nifedipine | 20 mg b.i.d., 15 d | 0* | Midazolam | 12) |
| Omeprazole | 20 mg q.d., 8 d | 0* | Nifedipine | 20) |
| Rosiglitazone | 8 mg q.d., 14 d | 0 | Ethinyl estradiol | 10) |
| 8 mg q.d., 14 d | 12 | Nifedipine | 24) | |
| 8 mg q.d., single dose | 0* | Simvastatin | 12) |
*% of AUC decrease was assumed to be 0 because the AUC of substrates in the presence of inducers was higher than that in the absence of inducers.
We evaluated CYP3A induction potential from in vitro data using the following methods, 7 of which are recommended by guidelines.
Fold-Change MethodWe calculated the fold increase in CYP3A mRNA levels using the following equation (eq. 5) assuming that the investigated inducer concentrations reflect the Cmax,u in humans.
 | (5) |
We defined negative CYP induction as having an mRNA increase of less than 2-fold and less than 20% of that by rifampicin (EmaxRIF), a positive control for the CYP3A inducer (eq. 6).
 | (6) |
We calculated the AUC ratio of substrate drugs in the presence and absence of inducers (R) using the following equation (eq. 7), where d represents the scaling factor and is usually assumed to be 1 in basic model. We defined positive CYP induction as having an R value of less than 0.9.
 | (7) |
We calculated the R value using the revised equation (eq. 8), which uses 10-fold Cmax,u instead of Cmax. We defined positive CYP induction when the R value was less than 0.8.
 | (8) |
We calculated the AUC ratio of substrate drugs in the presence and absence of inducers (AUCR) using the following equation (eq. 9). We defined positive CYP induction as having an AUCR value of less than 0.8.
 | (9) |
Bg and Bh refer to the intestinal and hepatic induction ratio of intrinsic clearance of substrates, respectively. They are represented by the equations 10 and 11, while [I]g and [I]h represent the maximum unbound inducer concentration in the intestine and the portal vein, respectively (eqs. 12, 13). Fa and ka refer to the fraction absorbed after oral administration and the first order absorption rate constant, respectively, which were assumed to be 1 and 6/h according to guidelines. Qen and Qh refer to enterocyte blood flow (18 L/h) and hepatic blood flow (97 L/h), respectively, according to guidelines. The blood-to-plasma concentration ratio (RB) of inducers was assumed to be 1. The scaling factor d was calculated by the geometric mean-fold error (GMFE) of the predicted value through linear weighted least-squares regression (eq. 14) for each batch of cells. We also used a scaling factor of 1.
 | (10) |
 | (11) |
 | (12) |
 | (13) |
 | (14) |
We calculated a RIS values of each inducer using the following equation (eq. 15) and plotted them against the AUC decrease of substrates in clinical studies to obtain the calibration curves for individual hepatocyte lots. We defined positive CYP induction as when RIS was larger than cut-off values at which AUC decrease of substrate drugs exceeds 20% in clinical studies.
 | (15) |
The calibration curves were fitted to a three-parameter sigmoid function (eq. 16) using non-linear least-squares regression and a programming solver (Microsoft Excel 2013), where A is the maximum % of AUC decrease, B is the RIS value achieving 50% of AUC decrease, and γ is the slope of the curve.10)
 | (16) |
We defined positive CYP induction when Cmax,u/EC50 was more than cut-off values at which AUC decrease of substrate drugs exceeds 20% in clinical studies. The calibration curves were obtained in the same way as in the RIS method (eq. 17).
 | (17) |
We defined positive CYP induction when AUCu/EC50 was more than cut-off values at which AUC decrease of substrate drugs exceeds 20% in clinical studies. The calibration curves were obtained in the same way as in the RIS method (eq. 18).
 | (18) |
We calculated the F2 value using the following equation (eq. 19) according to a previous report,16) while a Hill coefficient was set at 1 because the calibration curve could not be obtained.
 | (19) |
We defined positive CYP induction as AUC/F2 was more than cut-off values at which AUC decrease of substrate drugs exceeds 20% in clinical studies. The calibration curves were fitted to the following equation (eq. 20).16)
 | (20) |
Theoretically, the RF values are calculated from the ratio of IDLC of the standard inducer to that of a compound, where the IDLC value is determined using the following equation (eq. 21).15)
 | (21) |
C represents the lowest observed inducer concentration to show an induction response of more than 3-fold S.D. from the control level and I represents the observed fold induction at C.
The RF method assumes that Emax values of CYP3A induction are the same between different inducers and the induction response curves shift parallel depending on the EC50 values. Thus, the RF values estimated with the standard inducer, phenobarbital (RFPB), can be calculated using the following equation (eq. 22),15) where IDLCPB and EC50PB represent the IDLC and EC50 values of phenobarbital in each hepatocyte lot. We used in vitro EC50 values collected from literature to calculate RFPB in this study. We chose phenobarbital rather than rifampicin as a standard inducer for calculating RF value because the variability of the intrinsic induction activity (Emax/EC50) of phenobarbital was lower than rifampicin (Table 3).
We defined positive and negative CYP induction as when the multiplied value of Css,u and RFPB (eq. 23) was above the cut-off value of 5 mg rifampicin (Css,u_5RIF × RFPB_5RIF) and less than the cut-off value of 40 mg nifedipine (Css,u_40NIF × RFPB_40NIF). We set these cut-off values because they can be easily used for each hepatocyte lot compared to the first and last data points above and below the 20% decrease in the AUC of substrates (the upper and lower thresholds) as described previously.15) In the previous report, the different simple thresholds (30 mg phenobarbital for the upper threshold and 40 mg nifedipine for the lower threshold) were suggested because they used a dataset from different clinical induction studies; we used data on only the plasma AUC decrease of CYP3A substrates in this study, but the previous study also used data on the increase in urinary excretion of 6β-hydroxycortisol, the endogenous probe substrate. Thus, for phenobarbital, only a 75 mg dosing study was included in the present dataset where 15, 30, and 100 mg dosing studies were included in the previous dataset. Based on our chosen dataset, we reset the simple positive cut-off value near the upper threshold, 5 mg rifampicin, instead of 30 mg phenobarbital.
 | (22) |
 | (23) |
The incidence of false-positive and false-negative predictions for each clinical induction study were calculated as described below:
 | (24) |
 | (25) |
To equally evaluate each method, the incidence was calculated excluding rifampicin from the clinical study data because the fold-change method uses rifampicin as a positive control.
Emax and EC50 values of 9 inducers and non-inducers were collected from various literature and plotted in Fig. 1. Large inter-batch variability was observed in human hepatocytes; difference in Emax and EC50 values were 7.0 to 46-fold and 6.2 to 79-fold, respectively (Table 3). This is consistent with previous reports.30) On the other hand, mostly comparable Emax and EC50 values were observed in different lots of HepaRG, HepatoCells and Fa2N4. Due to the large variability in Emax and EC50, calculated assessment values also reflected variability between hepatocyte batches.

Open circles, red circles, green triangles and black rhombuses represent the data of distinct batches of hepatocytes, HepaRG cells, HepatoCells, and Fa2N4, respectively.
| Inducer | N | Mean | Median | CV% | Fold difference between the minimum and the maximum values | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Emax/EC50 | Emax | EC50 | Emax/EC50 | Emax | EC50 | Emax/EC50 | Emax | EC50 | Emax/EC50 | Emax | EC50 | ||
| Rifampicin | 22 | 69 | 33 | 0.81 | 41 | 21 | 0.56 | 120 | 98 | 91 | 65 | 38 | 29 |
| Phenobarbital | 19 | 0.10 | 25 | 512 | 0.07 | 18 | 239 | 92 | 85 | 214 | 88 | 15 | 60 |
| Carbamazepine | 22 | 0.46 | 14 | 45 | 0.26 | 11 | 36 | 140 | 63 | 65 | 45 | 9.1 | 26 |
| Phenytoin | 20 | 0.55 | 12 | 43 | 0.40 | 9.6 | 25 | 93 | 73 | 148 | 44 | 12 | 79 |
| Rifapentine | 6 | 42 | 44 | 1.3 | 37 | 24 | 1.1 | 78 | 93 | 65 | 10 | 8.1 | 6.2 |
| Pioglitazone | 17 | 1.0 | 14 | 16 | 0.72 | 7.7 | 12 | 88 | 114 | 80 | 11 | 32 | 22 |
| Nifedipine | 17 | 1.6 | 29 | 26 | 0.96 | 15 | 14 | 103 | 110 | 131 | 56 | 46 | 35 |
| Omeprazole | 5 | 0.40 | 6.8 | 18 | 0.43 | 6.1 | 14 | 33 | 71 | 70 | 2.5 | 7 | 6.5 |
| Rosiglitazone | 17 | 0.81 | 14 | 19 | 0.73 | 12 | 15 | 48 | 64 | 68 | 4.7 | 9.7 | 7.2 |
Large variability in hepatocytes might be due to the difference in the extent of induction response in the mRNA level of CYP3A, the quality of hepatocytes, the experimental conditions, and so on. There are some reports suggesting that the extent of induction response can be explained by the basal expression level of CYP3A30,31) or the subcellular concentration of inducers,32) which were only observed in limited number of hepatocyte batches. Thus, it is difficult to normalize the induction parameters using a single factor. As has been reported previously,30) the fold-change method, which normalizes the induction parameters to the positive control inducer, rifampicin, did not minimize inter-batch variability (Supplementary Table 2). As a result, although the DDI potential of negative compounds could be correctly evaluated in every batch of cells, some false-negative predictions were observed with the fold-change method (Fig. 2A). The reason for the failure to normalize using the induction response to rifampicin has not been clarified. It is possible that the subcellular concentration of rifampicin depends on polymorphism of OATP1B1 because it is a substrate for OATP1B133) and correlates less with the other inducers, and/or because the metabolic pathway primarily affected by rifampicin is very different from the other inducers. Minimal variability and similar Emax and EC50 values to hepatocytes were observed in cell lines and immortalized human hepatocytes, so it is possible they could substitute intact hepatocytes in predicting CYP3A-inducibility. However, Emax of some inducers in HepatoCells tended to be higher than in hepatocytes and other renewable cells (Fig. 1), which results in the overestimation of induction risk, leading to more false-positive predictions by the RIS approach as described previously.7)

(A) Different colored circles represent prediction data using each batch of cells. The black lines represent the 1.25-fold change in observed CYP3A mRNA level and the cut-off value of the predicted fold-induction (2). (B) Different colored circles represent prediction data using each batch of cells. The black lines represent the 0.8-fold change in observed AUCR and the cut-off value of the predicted R (0.9). (C) Different colored circles represent prediction data using each batch of cells. The black lines represent the 0.8-fold change in observed AUCR and the cut-off value of the predicted R (0.8). (D) Different colored circles represent prediction data using each batch of cells. The black lines represent the 0.8-fold change in observed AUCR and the cut-off value of the predicted AUCR (0.8). (E) Different colored circles represent prediction data using each batch of cells. The black lines represent the 0.8-fold change in observed AUCR and the cut-off value of the predicted AUCR (0.8).
We also implemented the basic model approaches recommended in the previous and revised guidance issued by FDA and MHLW. The previous basic model uses the total plasma concentrations of test compounds to predict the maximum induction potential. Thus, false-negative predictions were not observed in this assessment. However, almost every batch of cells showed false-positive predictions (Fig. 2B). By using 10-fold Cmax,u recommended by the revised guidance, the number of the false-positive predictions was considerably but not totally decreased. Some false-negative predictions were also observed for pioglitazone (Fig. 2C). It is possible that the direct use of Cmax,u further reduces false-positive predictions, but it might cause more false-negative predictions than the 10-fold Cmax,u.
In the mechanistic static model approach, the unbound plasma concentrations and intestinal concentrations were used to calculate the induction potential considering both hepatic and intestinal induction. We applied d values calculated from regression analysis for each hepatocyte and also the uniform value of 1. Using a d value of 1, risk assessment of positive compounds was almost completely successful (1 false-negative for phenobarbital) but there were lot of false-positive predictions, similar to the Basic model (Fig. 2D). When d values were calculated for respective hepatocytes, the number of false-positive predictions decreased, but the false-negative predictions increased (Fig. 2E).
To minimize the variability of induction responses in different batches of hepatocytes, the cut-off values for correlation approaches (RIS, Cmax,u/EC50, AUCu/EC50, AUC/F2 and RF methods) were set according to the calibration curve using data on known CYP3A inducers for each hepatocyte batch. For RIS, Cmax,u/EC50, AUCu/EC50, AUC/F2 methods, the calibration curve could not be obtained for some batches because the number of Emax and EC50 values for non-inducers were not sufficiently obtained. In such cases, we performed prediction using only the data with which the calibration curve could be obtained. As a result, correlation approaches showed fewer false predictions compared with the direct approaches that used the uniform cut-off values (Figs. 3, 4, Supplementary Figs. 1–5). The AUCu/EC50 and RF method which use the AUCu of the inducers were especially able to more adequately predict the induction risk: just 3 false positives out of 259 predictions in the AUCu/EC50 method and 5 false negatives out of 280 predictions in the RF method. On the other hand, RIS and Cmax,u/EC50, which use the Cmax,u of the inducers, or AUC/F2, which uses the total AUC of inducers showed relatively more false predictions: 21 and 19 false positives out of 251 predictions in the RIS and Cmax,u/EC50 methods, and 31 false positives and 2 false negatives out of 272 predictions in the AUC/F2 method. Better prediction using AUCu/EC50 over Cmax,u/EC50 was also reported elsewhere.1) This might be because the use of Cmax,u of inducers tends to overestimate the CYP3A inducibility at steady state more than the Css,u of inducers, which are proportional to AUCu.

(A) The black lines represent the 0.8-fold change in observed AUCR and the cut-off value of the predicted AUCu/EC50. (B) The black line represents the 0.8-fold change in observed AUCR. The red and green lines represent strong and weak induction thresholds, respectively.

Open and closed bars represent the percentage of false-positive and false-negative predictions, respectively. The number of false-positive, false-negative, and total predictions for each method is indicated above the open bars, below the closed bars, and below the graph, respectively.
With the RF method, because the intermediate region includes false-positive and false-negative compounds, the number of false predictions might be minimized compared with correlation methods that use a single threshold line. Predicting exact CYP3A inducibility is difficult, especially for moderate inducers. A previous report12) suggested that a Cmax,u/EC50 of 0.001 and 0.01 can be used as cut-off values for weak, moderate, and strong in vivo induction because of uncertainty in predicting the DDI of moderate inducers. However, the applicability of these cut-off values was only confirmed for the dataset obtained in three lots of hepatocytes in one laboratory. We set simpler cut-off values, nifedipine 40 mg and rifampicin 5 mg, to distinguish moderate compounds applicable to in vitro data obtained from diverse laboratories or systems. The RF and AUC/F2 methods resulted in a few false-negative predictions for pioglitazone, while the other correlation methods showed no false-negative prediction. The extent of induction by pioglitazone is relatively weak and differs based on the probe substrates used to calculate the induction level. We used data showing a 26% decrease in AUC of midazolam, the exogenous substrate. However, according to the urinary excretion data for 6β-hydroxycortisol, the endogenous probe substrate, the increase was less than 1.25-fold, which corresponds to a 20% decrease in the AUC of substrates.3–5) To avoid this false-negative prediction, the threshold for weak induction potential should be lowered for the RF and AUC/F2 methods. The in vivo induction potency of pioglitazone is low, however, so the threshold could be set according to the therapeutic range of possible co-administered substrate drugs.
When using the mean and median EC50 and Emax values for 9 inducers in the different batches of hepatocytes shown in Table 3, the correlation methods also achieved good predictions (Supplementary Table 4). This indicates that the correlation methods can accurately predict CYP3A induction by using the mean and median induction parameters of each drug.
| Method | Required parameter | Equations | Advantages | Disadvantages |
|---|---|---|---|---|
| Fold-change | Fold increase in CYP3A mRNA level of the compound and rifampicin at the expected in vivo concentration. | fold increase in CYP3A mRNA < 2 and <0.2→NEG | Only few parameters are required. Uniform threshold can be used. | Does not consider variability of hepatocytes. Prone to false-negatives. Cannnot be applied to compounds that both induce and inhibit CYP3A. Cannot be applied to poorly soluble and/or cell toxic compounds for which Emax and EC50 cannot be determined. |
| Basic model | In vitro Emax and EC50, and in vivo Cmax of the compound. |  | Only few parameters are required. Uniform threshold can be used. | Does not consider variability of hepatocytes. Prone to false-positives. Cannnot be applied to compounds that both induce and inhibit CYP3A. Cannot be applied to poorly soluble and/or cell toxic compounds for which Emax and EC50 cannot be determined. |
| Basic model using 10 × Cmax,u | In vitro fu, Emax, and EC50, and in vivo Cmax of the compound. |  | Only few parameters are required. Uniform threshold can be used. Fewer false-positives than Basic model using total Cmax. | Does not consider variability of hepatocytes. Cannnot be applied to compounds that both induce and inhibit CYP3A. Cannot be applied to poorly soluble and/or cell toxic compounds for which Emax and EC50 cannot be determined. |
| MS model (d = 1) | In vitro fu, Emax, and EC50 and in vivo Cmax of the compound and calibration compounds. In vivo fm and Fg of a substrate. |  | Uniform threshold can be used. Can consider induction both in liver and intestine. Can estimate proper induction by using fm and Fg of substrates. Can be applied for compounds both induce and inhibit CYP3A. | Does not consider variability of hepatocytes. Needs various in vitro and in vivo parameters. Prone to false-positives. Cannot be applied to poorly soluble and/or cell toxic compounds for which Emax and EC50 cannot be determined. |
| MS model | In vitro fu, Emax, and EC50 and in vivo Cmax of the compound. In vivo fm and Fg of a substrate. |  | Uniform threshold can be used. Can consider induction both in liver and intestine. Can estimate proper induction by using fm and Fg of substrates. Can be applied for compounds that both induce and inhibit CYP3A. Can include in vitro–in vivo scaling factor using calibration compounds. | Needs various in vitro and in vivo parameters. Needs many experiments to obtain calibration curves for each hepatocyte. Prone to false-positives. Cannot be applied to poorly soluble and/or cell toxic compounds for which Emax and EC50 cannot be determined. |
| RIS | In vitro fu, Emax, and EC50 and in vivo Cmax of the compound and calibration compounds. | 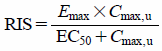  | Can minimize the effect of hepatocyte variability. Better correlation with clinical DDI compared with direct methods. | Need many experiments to obtain calibration curves for each hepatocyte and each laboratory because the standard set of known inducers for calibration is not specified. Cannnot be applied to compounds that both induce and inhibit CYP3A. Cannot be applied to poorly soluble and/or cell toxic compounds for which Emax and EC50 cannot be determined. |
| Cmax,u/EC50 | In vitro fu and EC50 and in vivo Cmax of the compound and calibration compounds. |  | Can minimize the effect of hepatocyte variability. Better correlation with clinical DDI compared with direct methods. | |
| AUCu/EC50 | In vitro fu and EC50 and in vivo AUC of the compound and calibration compounds. |  | Can minimize the effect of hepatocyte variability. Better correlation with clinical DDI compared with direct methods. Better prediction can be obtained by using the unbound AUC. | |
| AUC/F2 | In vitro F2 and in vivo AUC of the compound and calibration compounds. |  | Can minimize the effect of hepatocyte variability. Better correlation with clinical DDI compared with direct methods. Can reduce experiments because Emax and EC50 are not needed. | Need many experiments to obtain calibration curves for each hepatocyte and each laboratory because the standard set of known inducers for calibration is not specified. Cannnot be applied to compounds that both induce and inhibit CYP3A. Can overestimate inducibility of compounds with relatively lower induction responses. |
| RF | In vitro fu and IDLC and in vivo AUC of the compound and calibration compounds. |  Css,u corresponding to phenobarbital = Css,u × RFPB >Css,u_5RIF × RFPB_5RIF→POS <Css,u_40NIF × RFPB_40NIF→NEG | Can minimize the effect of hepatocyte variability. Better correlation with clinical DDI compared with direct methods. Can reduce experiments because Emax and EC50 are not needed and the full calibration curve is not needed. Better prediction can be obtained by using the unbound AUC. | Cannnot be applied to compounds that both induce and inhibit CYP3A. Cannot predict the inducibility by moderate inducers classified in intermediate region. |
Of the correlation methods, the RF and AUC/F2 methods do not require Emax and EC50 values, so they can be adapted to evaluate new chemical entities with poor solubility and cell toxicity using only low concentrations. Increasingly, many new clinical entities fall into the “beyond the rule of five” space and tend to show poor solubility,34) so these methods might be especially helpful for evaluating the next generation of drug candidates. As reported previously,16) the AUC/F2 approach based on the induction in HepaRG cells showed the best prediction when using the total plasma AUC. In this study, false-positives were observed for some hepatocytes, which might be due to the use of total plasma AUC (Supplementary Fig. 4). However, a recent study showed that the fold-change method using F2 and 30-fold of Cmax,ss,u showed fewer false-positives than with 50-fold of Cmax,ss,u,30) suggesting that predictions may be improved by modifying the calculation method to include the F2 value. In any case, for those inducers with relatively lower induction responses, F2 concentrations might be included in the saturated response and not in the linear region of concentration-response curve. In such a case, F2 might be wrongly estimated.
There are some limitations in this study. We evaluated the predictability of clinical induction using only 9 inducers and non-inducers, none of which were inhibitors of CYP3A. When we account for the reversible and time-dependent inhibition effect, the mechanistic model might be better for predicting clinical DDIs. Also, we collected the induction data from only 30 clinical studies, so variability in clinical induction could not be sufficiently evaluated here. Furthermore, we did not use other reported values based on initial slope of the concentration-response curve,35) and predictability using these values should also be evaluated.
In conclusion, our results indicate that correlation methods can more robustly and appropriately predict the CYP3A induction of new chemical entities compared with the direct approaches recommended by institutional guidelines. The predictability of the AUCu/EC50 and RF method, both of which use the AUCu of the inducers, was better than the other correlation methods. By using these methods along with the other correlation approaches to assess CYP3A induction risk, we might be able to better determine whether a clinical induction study should be performed or not in the early stages of drug development.
The authors wish to thank Dr. Motohiro Kato for helpful discussion regarding this paper. The authors also thank Mr. Jacob Davis for his useful advice in the preparation and language editing of this paper.
H. Tsutsui, S. Kuramoto, and K. Ozeki are employees of Chugai Pharmaceutical Co., Ltd. There are no other conflicts of interest.
The online version of this article contains supplementary materials.