Abstract
Background:
Long QT syndrome 2 (LQT2) is caused by mutations in the human ether-a-go-go-related gene (hERG). Most of its mutations give rise to unstable hERG proteins degraded by the proteasome. Recently, carbachol was reported to stabilize the wild-type hERG-FLAG via activation of the muscarinic type 3 receptor (M3-mAChR). Its action on mutant hERG-FLAG, however, remains uninvestigated.
Methods and Results:
A novel mutant hERG-FLAG carried 2 mutations: an amino acid substitution G572S and an in-frame insertion D1037_V1038insGD. When expressed in HEK293 cells, this mutant hERG-FLAG was degraded by the proteasome and failed to be transported to the cell surface. Carbachol restored stability of the mutant hERG-FLAG and facilitated cell-surface expression. Carbachol activated PKC, augmented phosphorylation of heat shock factor 1 (HSF1) and enhanced expression of heat shock proteins (hsps), hsp70 and hsp90. Both a M3-mAChR antagonist, 4-DAMP, and a PKC inhibitor, bisindolylmaleimide, abolished carbachol-induced stabilization of the mutant hERG-FLAG.
Conclusions:
M3-mAChR activation leads to enhancement of hsp expression via PKC-dependent phosphorylation of HSF1, thereby stabilizing the mutant hERG-FLAG protein. Thus, M3-mAChR activators may have a therapeutic value for patients with LQT2. (Circ J 2016; 80: 2443–2452)
KCNH2 encodes the α subunit of the human ether-a-go-go-related gene (hERG) channel that elicits the rapidly activating delayed-rectifier potassium current (IKr) responsible for action potential (AP) repolarization.1
Its mutation causes long QT syndrome type 2 (LQT2), which is characterized by prolongation of the QTc interval in electrocardiogram, and a high incidence of lethal arrhythmias.2,3
Most of the mutant hERG-FLAG have trafficking defects that result in reduced protein expression on the plasma membrane (PM).4
Heat shock factor 1 (HSF1) is a transcription factor that regulates expression of heat shock proteins (hsps).5
Hsps play a pivotal role in maturation and assemble of hERG channels.6
It has been reported that both hsp70 and hsp90 could facilitate maturation of the wild-type (WT) hERG-FLAG, and that hsp70 could facilitate that of mutant hERG-FLAG as well.7,8
The parasympathetic nervous system exerts negative inotropic and chronotropic actions on the heart through activation of muscarinic acetylcholine receptors (mAChRs). Among the five mAChR subtypes, M1, M2, M3 and M5 are expressed in the heart.9
M2-mAChR is mainly expressed in the atrium; it decreases the level of cAMP and simultaneously activates acetylcholine-sensitive K+
channels, exerting a negative chronotropic action.10
M3-mAChR is mainly expressed in the ventricle; it shortens the AP duration (APD) of cardiomyocytes via enhancing K+
channel currents.11
In mice, overexpression of M3-mAChR in the heart reduced the incidence of ventricular arrhythmias, suggesting its role in the prevention of arrhythmias.12
M3-mAChR is coupled with Gq
and activates the phospholipase C (PLC)-inositol triphosphate (IP3)-diacylglycerol (DAG) signaling pathway in the heart,13
which consequently activates PKC.14
PKC phosphorylates HSF1, leading to increased expression of hsp70.15
It has been shown that a muscarinic receptor agonist, carbachol, induced PKC-mediated phosphorylation of an E3 ubiquitin ligase, Nedd4-2, thereby inhibited hERG internalization and increased its level on the cell surface.16
Maturation of hERG is regulated by the endoplasmic reticulum-associated degradation (ERAD).17
It remains unknown whether carbachol could modulate ERAD and the ubiquitin-proteasome pathway.
In the present study, we examined carbachol effects on a novel mutant hERG-FLAG and found that carbachol stabilized the mutant hERG-FLAG through M3-mAChR. This effect was mediated by phosphorylation of HSF1 by PKC and enhanced expressions of hsp70 and hsp90.
Methods
Genomic Analysis
This study was approved by the Ethical Committee of the Faculty of Medicine at Tottori University and conformed to the principles outlined in the Declaration of Helsinki. Informed consent for participation in this study was obtained from the subject who was a 15-year-old male. Genomic DNA was extracted from his peripheral blood by using a standard phenol-chloroform procedure. A 5.7-kb genomic DNA fragment was amplified by PCR with primers: 5’-CAGGAATTCAGGAGGAGGGTCTAGGAAGTCTTTGGGG-3’ and 5’-CCGAAGCTTCTGGGCTAGGAATGGAAGAAGGGGATCC-3’. The digested PCR-amplified fragments with EcoRI and HindIII were sub-cloned into plasmid pTAKN-2 (BioDynamics Laboratory). Resulting plasmids were sequenced with M13 (-21) and M13 reverse primers from both ends.
Expression Plasmid
cDNA encoding the WThERG was cloned in a mammalian expression vector, pcDNA3.1 (+) (Invitrogen, Carlsbad, CA, USA) and the FLAG tag was ligated at the carboxy terminus of the hERG cDNA. The mutant hERG-FLAG (G572S-D1037_V1038insGD) cDNA was generated by using a QuikChange site-directed mutagenesis kit (Stratagene).
Cell Culture and Establishment of HEK293 Cell Lines Stably Expressing WT and the Mutant hERG-FLAG
Cells were cultured in Dulbecco’s modified Eagle’s medium (D-MEM; Wako), supplemented with 10% fetal bovine serum (Nichirei Biosciences, Tokyo, Japan) and 0.5% penicillin-streptomycin G (Wako) at 37o
C in a 5% CO2
incubator. Each plasmid was transfected into HEK293 cells using lipofectamine 2000 (Invitrogen). Stable cell lines were generated by using the antibiotic, G418 (Wako), for selection (1 mg/ml) and maintenance (0.4 mg/ml).
Materials
A M3-mAChR antagonist, diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP), and an M2-mAChR antagonist, AF-DX 116 (11[[2-[(diethylamino)methyl]-1-piperidinyl]-acetyl]-5,11-dihydro6H-pyrido[2,3-b] [1,4]benzodiazepine-6-one), were from Abcam, and a PKC inhibitor, bisindolylmaleimide I hydrochloride (Bis-I), was from Adipogen. These inhibitors were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO in the culture medium was equal to or less than 0.01% (v/v). Carbamylcholine chloride (Carbachol) from Sigma and other drugs were dissolved in Milli-Q water.
Western Blotting
Cells were scraped into lysis buffer (PBS containing 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstain and 1 mmol/L phenylmethylsulfonyl fluoride) and lysed by sonication; insoluble materials were removed by centrifugation. Protein concentrations were determined with a protein assay kit (Bio-Rad). Ten μg of proteins were separated on SDS-PAGE and electrotransferred to a PVDF membrane (Immobilon-P; Millipore). Membranes were probed with antibodies against FLAG (Agilent Technologies), α-tubulin (Abcam), hsp70 (Enzo Life Sciences), hsp90 (Santa Cruz), Ser326-phosphorylated HSF1 (Abcam), HSF1 (Merck Millipore), M3-mAChR (Santa Cruz), M2-mAChR (Santa Cruz) and were developed using an ECL system (Amersham). Band intensities were quantified using Image J software.
Chase Assay
HEK293 cells stably expressing WT or the mutant hERG-FLAG were seeded in 6-well plates. Brefeldin A (Sigma) was administrated to cells for 12 h before carbachol treatment. After the addition of cycloheximide (60 μg/ml), protein extracts were prepared at the indicated times and subjected to anti-FLAG Western blotting.
Immunofluorescence
HEK293 cells were seeded on gelatin-coated coverslips and transfected with WT or the mutant hERG-FLAG plasmid, together with pDsRed2-ER (Clontech, Mountain View, CA, USA), pDsRed-Monomer-Golgi (Clontech) or pPM-mKeima-Red (BML, Tokyo, Japan). All the staining procedures were conducted at room temperature. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X-100 for 10 min, and blocked with 3% albumin in PBS for 30 min. Cells were incubated for 1 h with the anti-FLAG antibody (Agilent Technologies). Bound antibodies were visualized with AlexaFluor 488-conjugated mouse secondary antibody, and images were obtained by using a Bio-Rad MRC 1024 confocal microscope. To quantify hERG-FLAG signals, images were cropped with regard to the distribution of each marker protein (DsRed2-ER, DsRed-Monomer-Golgi or PM-mKeima-Red) using Photoshop CS3 software. Signal intensities in cropped areas were quantified by using Image J software.
Reporter Gene Assay
A plasmid encoding a luciferase gene driven by the hsp70 promoter (phsp70 firefly luciferase) was provided by A. Nakai. HEK293 cells expressing WT and the mutant hERG-FLAG were prepared in 96 wells at 70–80% confluencies. CMV-Renilla
luciferase plasmid (pCMV-Renilla
luciferase) (Promega, WI, USA) and phsp70 firefly luciferase were co-transfected into the cells. After 24 h of transfection, cells were treated with or without carbachol. The luciferase activity in the cell lysates was determined by using Dual-Glo®
(Promega), according to the manufacturer’s instructions. The results were expressed as the hsp70 luciferase activity normalized to
Renilla
luciferase activity.
Electrophysiological Recordings
The hERG channel currents (IhERG) corresponding to
IKr
were measured by whole-cell patch-clamp techniques with an Axopatch-200B amplifier (Molecular Devices). The extracellular solution contained (mmol/L): NaCl 140, KCl 4, CaCl2
1.8, MgCl2
0.53, NaH2PO4
0.33, glucose 5.5, and HEPES 5 (pH 7.4). The internal pipette solution contained (mmol/L): K-aspartate 100, KCl 20, CaCl2
1, Mg-ATP 5, EGTA 5, HEPES 5, and creatine phosphate dipotassium salt 5 (pH 7.2). Patch pipettes had a resistance of 2–4 MΩ. After rupture of the cell membrane, whole-cell membrane currents were recorded at 37℃ under the voltage-clamp mode. Series resistance (Rs) was determined by fitting a single exponential function to the capacitive current decay to estimate its time constant (τ) and the membrane capacitance (Cm); Rs
calculated with the equation Rs=τ/Cm
during the capacitive current cancellation ranged 2.2±0.4 MΩ with τ=98±8 μs and Cm=46±24 pF (n=5). After the Rs
compensation of 50–60%, the voltage errors arising from Rs
were estimated to be less than 5 mV. The membrane potential was not corrected for the liquid junction potential, which was estimated to be <10 mV. Holding potential was set at –80 mV and
IhERG
was evoked by 2-s depolarizing test pulses ranging from –50 to +50 mV (with a 10-mV increment) followed by repolarization to –60 mV for 6 s.18
A blocker selective for hERG channels, E4031 (Wako), was added at 10 μmol/L; E4031-sensitive currents were measured by digital subtraction of the current traces with and without E4031. The peak current amplitudes were determined during test pulses and plotted as functions of depolarizing pulses.
Computer Simulations of APs of Human Ventricular Myocytes (HVMs)
We simulated prolongation of APDs and generation of early afterdepolarization (EAD) in LQT2 HVMs with the mutant hERG using mathematical models developed by Kurata et al19
and O’Hara et al.20
A mid-myocardial (M) cell version of the Kurata et al model was developed on the basis of the transmural heterogeneity in densities of sarcolemmal ion channels, transporters and SR Ca2+
uptake/release rates, as summarized by O’Hara et al.20
We used the M cell versions of the two HVM models because they have larger
ICaL, as well as smaller
IKr
and
IKs, thus being much more vulnerable to EAD formation than the endocardial and epicardial versions.20
Whether the carbachol-induced modification of the mutant hERG current inhibits EAD formation in the LQT2 HVMs was tested, as shown in the supplementary material.
Statistical Analysis
All the data were presented as mean±SEM, and analyzed by using a Student’s
t-test or repeated measures analysis of variance (2-way ANOVA). The differences between groups were considered statistically significant at P<0.05.
Results
Characterization of the Mutant hERG-FLAG Channel Protein
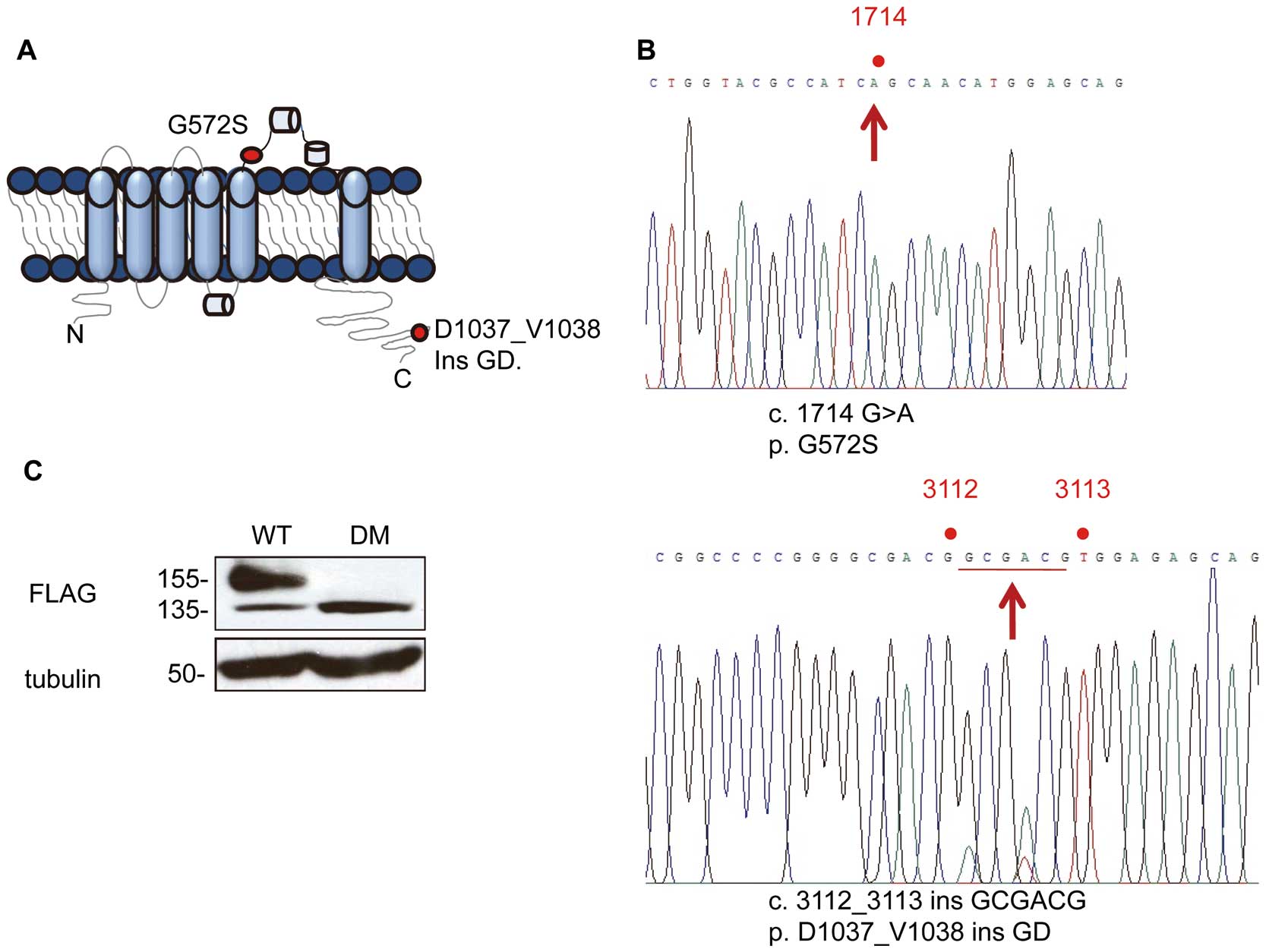
Two mutations of hERG were found in the genome of a LQT2 patient with a QTC
of 520 ms (Figure 1A). One causes the amino acid substitution G572S and the other causes the amino acid insertion D1037_V1038insGD (Figure 1B). Pedigree and ECGs of the patient and his family are shown in the
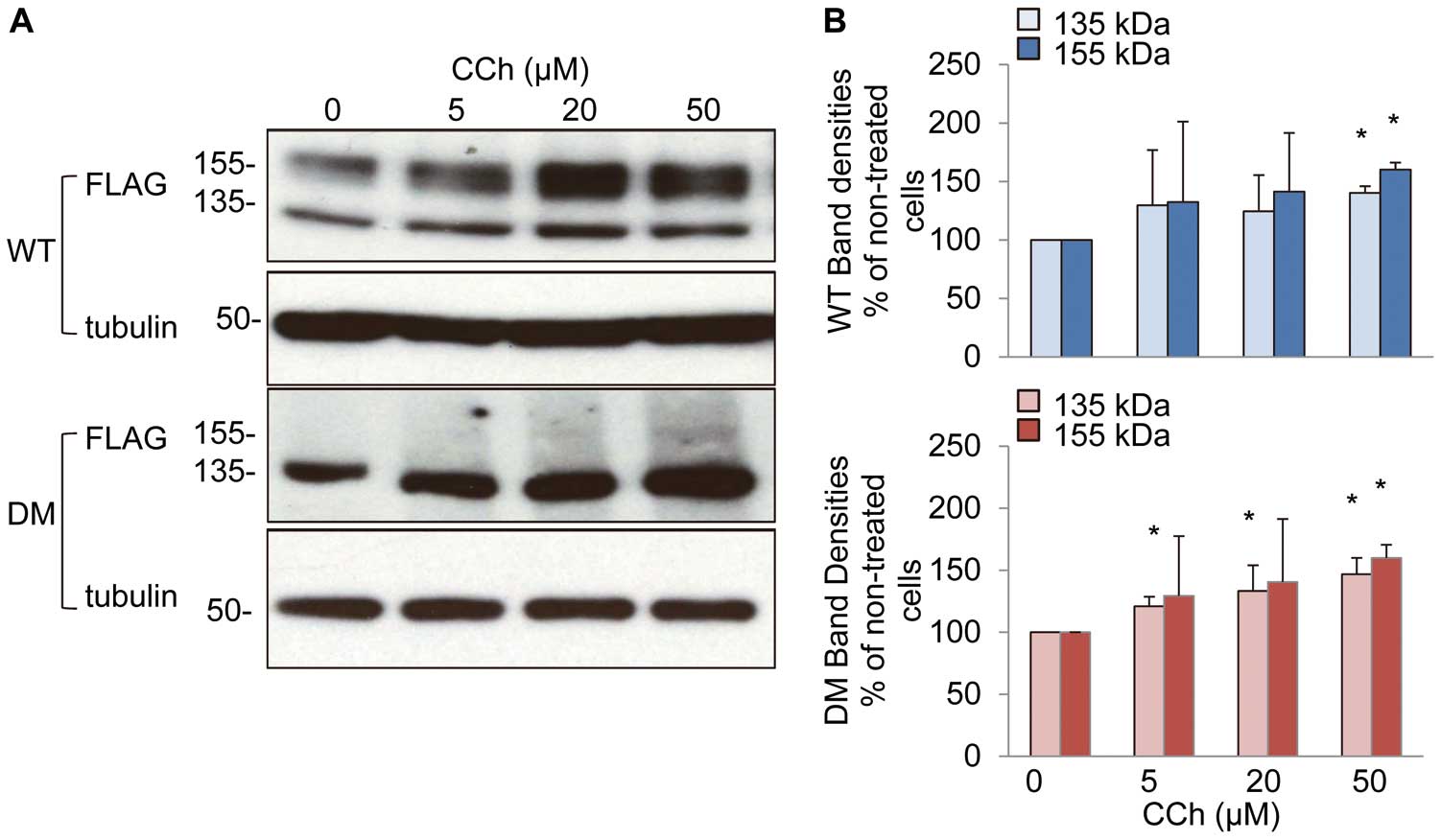
Figure S1. Next, we examined whether the 2 mutations beside in the same. Genomic DNA fragments that contained both mutation sites were amplified by PCR. Ten colonies were randomly chosen and the sub-cloned DNA fragments were sequenced. Sequencing of 7 colonies revealed both mutations resided in a genomic fragment while 3 colonies represented the WT hERG sequence, indicating that the patient had a double mutant in the single allele. When expressed in HEK293 cells, WThERG-FLAG gave a 155-kDa mature and a 135-kDa immature forms, whereas the mutant hERG-FLAG exclusively remained in its immature form (Figure 1C).
It has been reported that the G572S single mutant exerted a dominant negative effect.21
Therefore, we examined whether the double mutant has the same effect by co-expression as reported previously. Co-expression of G572S resulted in a drastic decrease in the E-4031-sensitive currents. This effect was observed in the double mutant and was barely observed with the insertion. Thus, the double mutant also has the dominant-negative effect, albeit much less than G572S (Figure S2).
Carbachol Facilitates Maturation of the Mutant hERG-FLAG
We examined carbachol effects on hERG maturation in HEK293 cells stably expressing hERG-FLAG. Carbachol caused dose-dependent increases of both mature and immature forms of the mutant hERG-FLAG (DM), as well as WT (Figures 2A,B). Time-course analysis showed that a 2 h treatment with carbachol at 50 μmol/L was enough to induce the maximum effect (Figures S3A,B). Carbachol also increased the mature form of another mutant hERG G601S-FLAG (Figure S4).
Immunofluorescence showed that the signals of both the WT and the mutant hERG-FLAG co-localized with those of the ER (#3, 12) (Figure 3A). The signals of the mutant hERG-FLAG were slightly decreased in the Golgi apparatus (#15) and were absent on the PM (#18). Treatment with carbachol significantly increased the signals of the mutant hERG-FLAG in the Golgi apparatus (#24) as well as on the PM (#27). The changes in immunoreactivities were confirmed by quantification of the signal activities (Figure 3B).
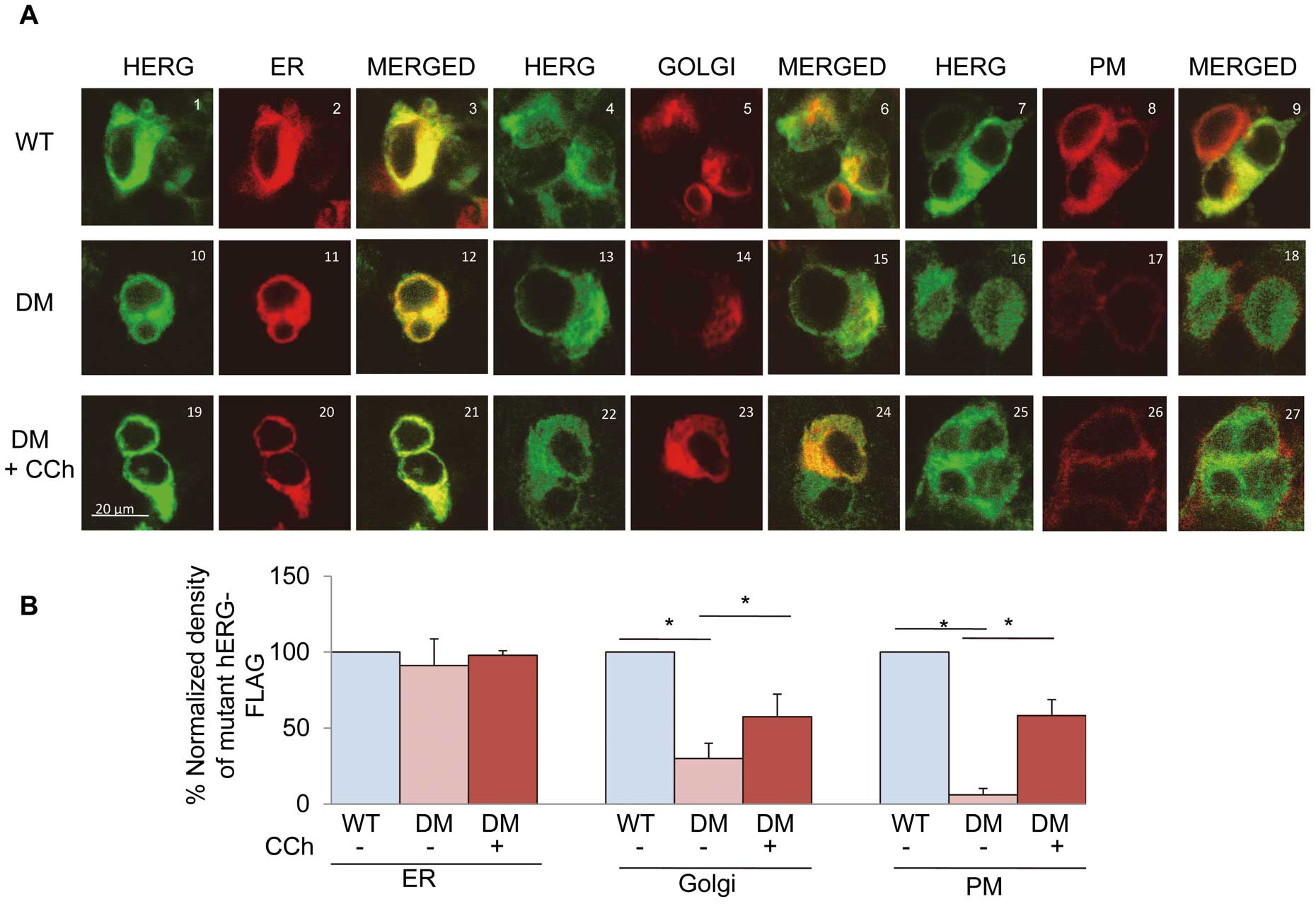
Figure 4A
shows the original traces of E4031-sensitive currents recorded in HEK293 cells stably expressing the WT or mutant hERG-FLAG. Depolarizing pulses activated time-dependent outward currents that corresponded to
IKr
in cells expressing WThERG-FLAG. Cells expressing the mutant hERG-FLAG showed substantially reduced E4031-sensitive currents. At +10 mV, the peak currents were decreased by ~80% compared with the currents in cells expressing the WT. Treatment with carbachol increased the currents (Figure 4B). These results suggested that carbachol enhanced maturation of the mutant hERG-FLAG proteins, which are then transported to the cell surface.

Carbachol appeared to facilitate maturation of the mutant hERG-FLAG. To see whether carbachol could affect ERAD of the mutant hERG-FLAG, we examined the effects of carbachol in the presence of brefeldin A, which blocks protein transport from the ER to the Golgi apparatus, and cycloheximide, which blocks the protein synthesis. Degradation of the mutant hERG-FLAG protein was faster than that of WThERG-FLAG, and carbachol suppressed its degradation (Figures 5A,B). Thus, carbachol could stabilize the mutant hERG-FLAG by suppressing its ERAD.
Carbachol Facilitates Maturation of the Mutant hERG-FLAG Through Activation of M3-mAChR
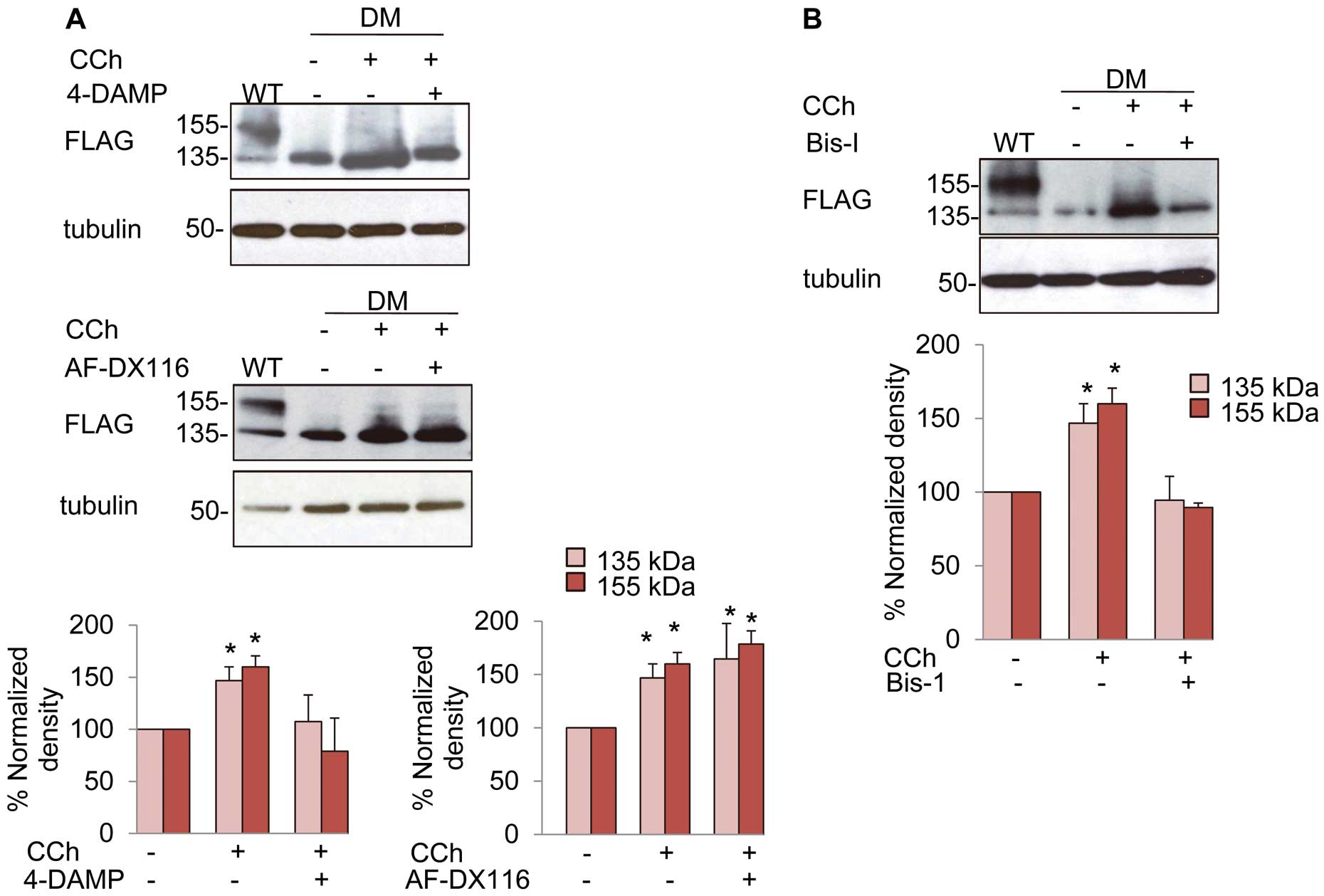
To identify the mAChR subtype responsible for the carbachol effects, we used a M3-mAChR antagonist, 4-DAMP, and a M2-mAChR antagonist, AF-DX 116, because HEK293 cells expressed both M2-mAChR and M3-mAChR (Figure S5A). 4-DAMP, but not AF-DX 116, abolished the effect of carbachol on the protein levels of the mutant hERG-FLAG (Figure 6A) as well as WThERG-FLAG (Figures S5B,C), indicating the involvement of M3- but not M2-mAChR.
In order to examine whether the PKC pathway is involved in the action of carbachol, we studied the effects of a PKC inhibitor, Bis-I, on the carbachol-induced maturation of the mutant hERG-FLAG. Bis-I at 3 μmol/L abolished the carbachol effects on the mutant hERG-FLAG (Figure 6B) as well as on WThERG-FLAG (Figure S6A).
Carbachol Facilitates Maturation of the Mutant hERG-FLAG Through Phosphorylation of HSF1
Both Hsp70 and hsp90 facilitate maturation of the WThERG-FLAG and mutant hERG-FLAG.7,8
Expressions of both hsps are regulated by HSF1, which is one of the downstream effectors of PKC.5,15
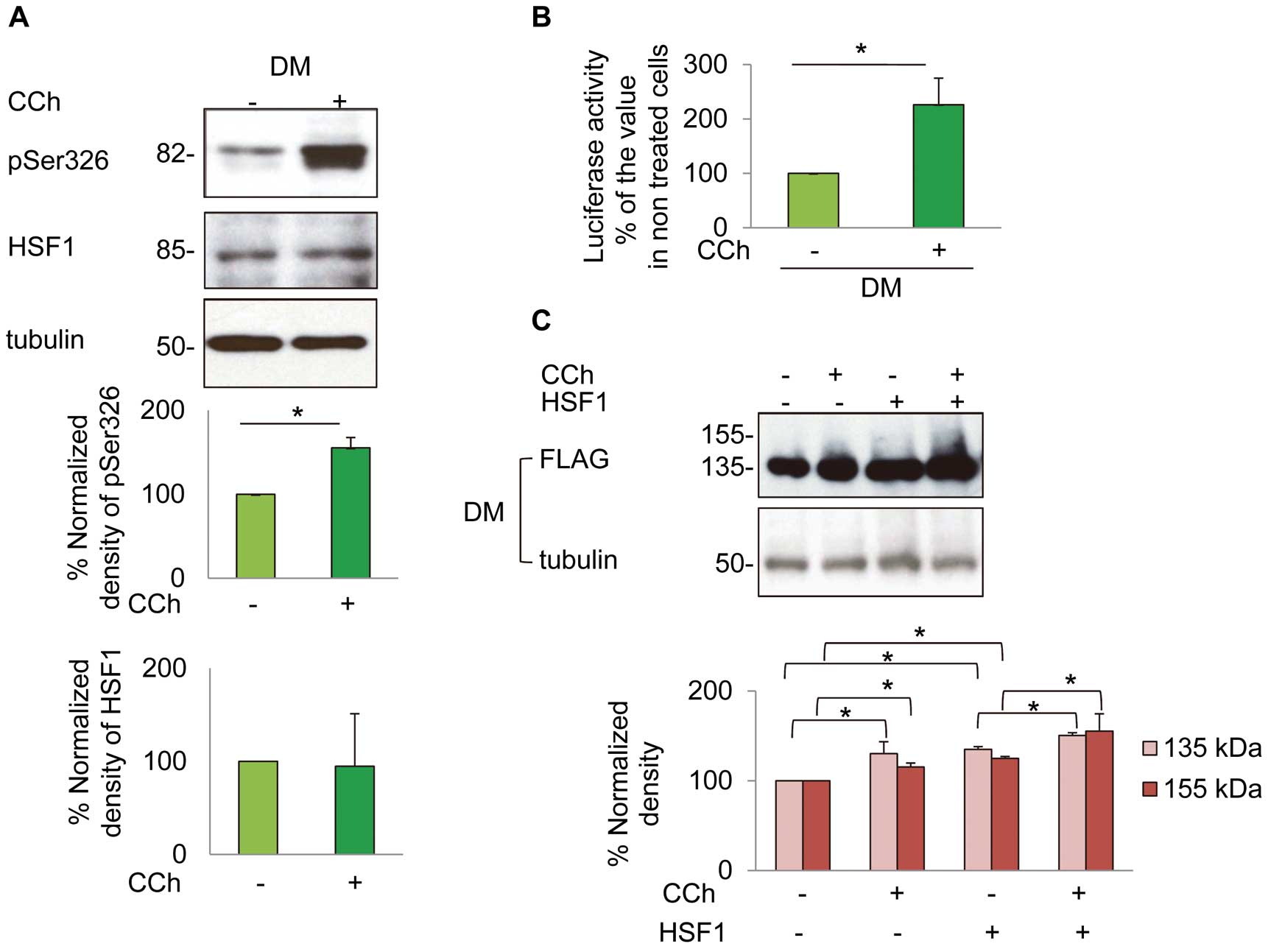
To examine whether carbachol could induce phosphorylation of HSF1, we used an antibody that recognizes HSF1 with a phosphorylated serine residue at aa 326 (pSer326). Treatment with carbachol increased the level of pSer326 in cells expressing the mutant hERG-FLAG without changes in the total protein level of HSF1 (Figure 7A).
To confirm the role of the HSF1-hsp pathway, we examined whether carbachol could increase the transcription of hsp70 by using a reporter gene assay. Treatment with carbachol increased transcription of the hsp70 reporter gene (Figure 7B) and enhanced the protein levels of hsp70 and hsp90 (Figures 8A,B). Moreover, carbachol could further facilitate maturation of the mutant hERG-FLAG when cells were co-transfected with HSF1 as well as the WThERG-FLAG (Figure 7C;
Figure S6B).
In accordance with these findings, inhibition of the M3-mAChR-PKC pathway by 4-DAMP (1 μmol/L) or Bis-I (3 μmol/L) abolished carbachol-induced increases of hsp70 and hsp90, as well as pSer326 (Figures 8A,B).
Discussion
We found novel double mutations of the hERG gene in a Japanese male LQT2 patient; one was an amino acid substitution, G572S, in the pore domain and the other was an inframe insertion of D1037_V1038insGD in the carboxyl terminus. The mutant hERG-FLAG was expressed solely as an immature form at 135 kDa. It was localized predominantly in the ER and was not expressed on the cell surface. Carbachol induced the mature form (155 kDa) of the mutant hERG-FLAG. We demonstrated that carbachol activated M3-mAchR to phosphoryilate HSF1 via activation of the PKC, resulting in activation of hsp family proteins and maturation of the mutant hERG-FLAG. Carbachol has been reported to increase the mature form of WThERG-FLAG via inhibition of its internalization, and the current study is the first report to demonstrate that the same drug could facilitate maturation of the mutant hERG-FLAG at the ER.
Muscarinic receptors mediate parasympathetic nerve actions on the heart. M1-, M2-, M3- and M5-mAChR have been reported to be expressed in the human heart, and each of them has different distribution and function.9
M3-mAChR is predominantly expressed in the ventricle and exerts negative inotropic and chronotropic effects. It also regulates cardiac AP repolarization and cell-to-cell communication.22
Two subtypes of muscarinic receptors, M2- and M3-mAChR, have been reported to be expressed in HEK293 cells.23
The M3-mAChR antagonist, 4-DAMP, but not the M2-mAChR antagonist, AF-DX116, abolished carbachol-induced maturation of the mutant hERG-FLAG, indicating the involvement of M3-mAChR. The role of PKC was evidenced by the Bis-I effect on carbachol-induced maturation of the mutant hERG-FLAG. Previous reports show that M3-mAChR could activate PKC through Gq
proteins.24
It has been reported that carbachol induces phosphorylation of Nedd4-2 via the PKC pathway through M3-mAChR, inhibits monoubiquitination of WThERG-FLAG and suppresses its internalization from the PM. This results in inhibition of its degradation in the endosome and an increase of channel function.16,25
We showed that carbachol facilitated maturation of the trafficking-deficient mutant hERG-FLAG via the PKC pathway through activation of M3-mAChR, and that this effect was mediated by phosphorylation of HSF1. HSF1 is activated by 2 steps. On the first step, HSF1 binds to HSEs and forms an oligomer; and on the second step, it is phosphorylated to exert a high transcriptional activity.5
The serine residue at position 326 in HSF1 has been identified as a dominant target for phosphorylation regulators.26
As shown in
Figure 7, carbachol increased phosphorylation of Ser326 in HSF1 and activated the promoter activity of hsp70. This subsequently increased protein levels of hsp70 and hsp90. Overexpression of HSF1 enhanced the maturation of the hERG protein, providing further evidence for the role of HSF1.
Our novel mutant hERG-FLAG yielded very small currents and had a dominant negative effect. The currents mediated by the mutant hERG-FLAG appear to be much faster than that of the WThERG-FLAG, suggesting that the mutant channel has distinct gating properties. Therefore, carbachol may not be beneficial in shortening APDs or inhibiting EAD formation, especially if the mutant proteins harbor severe gating abnormalities. Thus, we tested whether the carbachol-induced increase in the mutant channels could lead to APD shortening by computer simulation using the Kurata et al and O’Hara et al models for HVMs.19,20
Both models predicted that the carbachol-induced increase in the mutant hERG-FLAG could yield APD shortening in HVMs expressing the mutant hERG (Figure S7). Thus, the enhancement of the mutant hERG-FLAG current by carbachol could be beneficial for certain LQT2 patients.
There are some limitations in our study, which are as follows: (1) only the proband exhibited the prominent prolongation of QTc, suggesting that the G572S-D1037_V1038insGD mutation in 1 allele may be a de novo mutation. Unfortunately, other family members’ genomic information on the hERG gene was lacking because of their refusal to our proposal for genetic analysis; and (2) we used the HEK293 cells stably expressing hERG channels to examine the effects of carbachol on the maturation of the mutant hERG-FLAG, because the HEK293 cells are well known to be capable of characterizing cardiotoxic agents that are involved in prolongation of QT intervals and display biophysical and pharmacological properties similar to those in native cardiomyocytes. However, the heterologous expression system is not identical to native cardiomyocytes in terms of trafficking and degradation of the hERG protein. Cell-specific post-transcriptional and post-translational modifications are very important for channel functions, as suggested recently.27
Carbachol facilitated the maturation of endogenous ERG in HL-1 cells as well as the mutant hERG-FLAG in transfected HL-1 cells (Figure S8). Nevertheless, further experiments using iPS cell-derived cardiomyocytes originated from LQT2 patients will be necessary.
Acknowledgments / Conflicts of Interest
None.
Supplementary Files
Supplementary File 1
Figure S1.
Family history and ECG recordings of the long QT syndrome 2 (LQT2) patient and his family members.
Figure S2.
Dominant negative effects of the double mutant hERG-FLAG.
Figure S3.
Time-dependent effect of carbachol (CCh) treatment duration on the maturation of WThERG-FLAG and mutant hERG-FLAG (A,B).
Figure S4.
Effects of carbachol on another mutant hERG, G601S hERG-FLAG, expressed in HEK293 cells.
Figure S5.
Expression of M2 and M3 receptors on the HEK293 cells stably expressing WThERG-FLAG (wild-type; WT) and mutant hERG-FLAG (double mutation; DM) (A).
Figure S6.
(A) Effects of the PKC inhibitor, bisindolylmaleimide (Bis-I), on WThERG-FLAG expression.
Figure S7.
Simulated action potential (AP) and delayed rectifier potassium current (IKr) behaviors of the Kurata et al (A) and O’Hara et al (B) models for human ventricular myocytes (HVMs) expressing WThERG (black) or the mutant human ether-a-go-go gene (hERG) in the absence (red) and presence of carbachol (blue).
Figure S8.
Effects of carbachol (CCh) on expressions of the mouse ERG (mERG) and the expressed mutant hERG-FLAG in HL-1 mouse cardiomyocytes.
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-16-0712
References
- 1.
Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995; 81: 299–307.
- 2.
Wilde A, Tan HL. Inherited arrhythmia syndromes. Circ J 2007; 71: 12–19.
- 3.
Nishizaki M, Hiraoka M. Gene mutations associated with atrioventricular block complicated by long QT syndrome. Circ J 2010; 74: 2546–2547.
- 4.
Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 2006; 113: 365–373.
- 5.
Sorger PK. Heat shock factor and the heat shock response. Cell 1991; 65: 363–366.
- 6.
Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel hERG. Circ Res 2003; 92: e87–e100, doi:10.1161/01.RES.0000079028.31393.15.
- 7.
Li P, Ninomiya H, Kurata Y, Kato M, Miake J, Yamamoto Y, et al. Reciprocal control of hERG stability by Hsp70 and Hsc70 with implication for restoration of LQT2 mutant stability. Circ Res 2011; 108: 458–468.
- 8.
Iwai C, Li P, Kurata Y, Hoshikawa Y, Morikawa K, Maharani N, et al. Hsp90 prevents interaction between CHIP and HERG proteins to facilitate maturation of wild-type and mutant HERG proteins. Cardiovasc Res 2013; 100: 520–528.
- 9.
Wang H, Han H, Zhang L, Shi H, Schram G, Nattel S, et al. Expression of multiple subtypes of muscarinic receptors and cellular distribution in the human heart. Mol Pharmacol 2001; 59: 1029–1036.
- 10.
Kitazawa T, Teraoka H, Harada N, Ochi K, Nakamura T, Asakawa K, et al. Regulation of heart contractility by M2 and M3 muscarinic receptors: Functional studies using muscarinic receptor knockout mouse. In: Myslivecek J, Jakubik J, editors. Muscarinic receptor: From structure to animal models. New York: Springer, 2016, 235–259.
- 11.
Shi H, Wang H, Wang Z. Identification and characterization of multiple subtypes of muscarinic acetylcholine receptors and their physiological functions in canine hearts. Mol Pharmacol 1999; 55: 497–507.
- 12.
Liu Y, Sun L, Pan Z, Bai Y, Wang N, Zhao J, et al. Overexpression of M3 muscarinic receptor is a novel strategy for preventing sudden cardiac death in transgenic mice. Mol Med 2011; 17: 1179–1187.
- 13.
Mathes PW, Leineweber K, Wangemann T, Silber RE, Brodde OE. Existence of functional M3-muscarinic receptors in the human heart. Naunyn Scmiedebergs Arch Pharmacol 2003; 368: 316–319.
- 14.
Shi H, Wang H, Yang B, Xu D, Wang Z. The M3 receptor-mediated K+ current (I KM3) a Gq protein-coupled K+ channel. J Biol Chem 2004; 279: 21774–21778.
- 15.
Yamanaka K, Takahashi N, Matsumoto H, Ohnishi T, Kaneda K, Yoshimatsu H, et al. Role of protein kinase C in geranylgeranylacetone-induced expression of heat-shock protein 72 and cardioprotection in the rat heart. J Mol Cell Cardiol 2003; 35: 785–794.
- 16.
Wang T, Hogan-Cann A, Kang Y, Cui Z, Guo J, Yang T, et al. Muscarinic receptor activation increases hERG channel expression through phosphorylation of ubiquitin ligase Nedd4-2. Mol Pharmacol 2014; 85: 877–886.
- 17.
Nishikawa SI, Brodsky JL, Nakatsukasa K. Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J Biochem 2005; 137: 551–555.
- 18.
Sakaguchi T, Itoh H, Ding WG, Tsuji K, Nagaoka I, Oka Y, et al. Hydroxyzine, a first generation H1-receptor antagonist, inhibits human ether-a-go-go-related gene (HERG) current and causes syncope in a patient with the HERG mutation. J Pharmacol Sci 2008; 108: 462–471.
- 19.
Kurata Y, Hisatome I, Matsuda H, Shibamoto T. Dynamical mechanisms of pacemaker generation in IK1-downregulated human ventricular myocytes: Insights from bifurcation analyses of a mathematical model. Biophys J 2005; 89: 2865–2887.
- 20.
O’Hara T, Virag L, Varro A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput Biol 2011; 7: e1002061, doi:10.1371/journal.pcbi.1002061.
- 21.
Zhao JT, Hill AP, Varghese A, Cooper AA, Swan H, Laitinen-Forsblom PJ, et al. Not all hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J Cardiovasc Electrophysiol 2009; 20: 923–930.
- 22.
Wang H, Lu Y, Wang Z. Function of cardiac M3 receptors. Auton Autacoid Pharmacol 2007; 27: 1–11.
- 23.
Kurian N, Hall CJ, Wilkinson GF, Sullivan M, Tobin AB, Willars GB. Full and partial agonists of muscarinic M3 receptors reveal single and oscillatory Ca2+ responses by β2-adrenoceptors. J Pharmacol Exp Ther 2009; 330: 502–512.
- 24.
Luo J, Busillo J, Benovic J. M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol Pharmacol 2008; 74: 338–347.
- 25.
Guo J, Wang T, Li X, Shallow H, Yang T, Li W, et al. Cell surface expression of human ether-a-go-go-related Gene (hERG) channels is regulated by caveolin-3 protein via the ubiquitin ligase Nedd4-2. J Biol Chem 2012; 287: 33132–33141.
- 26.
Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 2005; 6: 4.
- 27.
Solana J, Gamberi C, Mihaylova Y, Grosswendt S, Chen C, Lasko P, et al. The CCR4-NOT complex mediates deadenylation and degradation of stem cell mRNAs and promotes planarian stem cell differentiation. PLoS Genet 2013; 9: e1004003, doi:10.1371/journal.pgen.1004003.