Editorials
Long-Term Prognosis of Catecholaminergic Polymorphic Ventricular Tachycardia Patients With Ryanodine Receptor (RYR2) Mutations
2016 Volume 80 Issue 9 Pages 1892-1894


Details
2016 Volume 80 Issue 9 Pages 1892-1894
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a malignant inherited arrhythmia syndrome characterized by bidirectional or polymorphic ventricular arrhythmias under conditions of increased sympathetic activity in young patients without structural heart disease.1 The common autosomal-dominant form has been linked to mutations in the gene encoding the cardiac calcium-release channel (RYR2).2 To date at least 130 unique, almost exclusively missense, mutations in RYR2 have been reported.3 A less common but more severe autosomal-recessive form is caused by mutations in the gene encoding cardiac calsequestrin (CASQ2), the major calcium-binding protein in the sarcoplasmic reticulum (SR).4 Mutations in these genes have been identified in approximately 60% of patients diagnosed as CPVT,2 and result in inappropriate calcium leakage from the SR, leading to cytosolic calcium overload generating delayed afterdepolarizations, triggered activity, and ventricular arrhythmias, particularly under increased β-adrenergic tone. CPVT has become recognized as a significant cause of sudden cardiac death in children and young adults. In untreated patients 8-year overall arrhythmic event rates of 58% and fatal or non-fatal event rates of 25% have been documented.5 A history of aborted cardiac arrest, absence of β-blockers5, and carrying recessively inherited CASQ2 mutations6 have been reported as risk factors of arrhythmic events.
Article p 1907
In this issue of the Journal, Kawata and colleagues7 evaluate the recurrence rate of fatal cardiac events after initiation of appropriate medical therapy in 34 CPVT patients with RyR2 mutations. During 7.4 years of follow-up, 7 of the 34 patients developed fatal cardiac events, and among them, 6 patients (85.7%) were not compliant with exercise restriction or medication therapy. In other words, the majority of CPVT patients were well controlled by appropriate medical therapy, emphasizing the importance of drug compliance and advice to the patients and their families about not practicing (competitive) sports.
Figure 1 shows the molecular and signal targets of medical treatment for CPVT. Arguably, β-blockers are the cornerstone of drug therapy for reducing ventricular arrhythmias during exercise testing and preventing arrhythmic events.8 The first step in treating CPVT patients should be a β-blocker at the highest tolerable dose. Adding a calcium blocker to β-blocker could be better than β-blocker alone for preventing exercise-induced arrhythmias in CPVT.9 L-type calcium-channel blockers such as verapamil inhibit the calcium-induced calcium release through RYR2 and also decrease the calcium content of the SR. Although the precise mechanism is not clear, calcium-channel blockers have been shown to be clinically effective in CPVT.9 Notably, the antiarrhythmic agent flecainide directly targets the molecular defect in CPVT by inhibiting premature calcium release and triggered beats.10 Flecainide is reported to reduce exercise-induced ventricular arrhythmias even in patients not controlled by β-blocker and/or calcium blocker therapy, and probably plays an irreplaceable role in the treatment of CPVT patients.11 In addition, dantrolene, a RYR2 channel inhibitor and effective in malignant hyperthermia, has been shown to prevent exercise- and epinephrine-induced ventricular arrhythmias in CPVT mouse models.12 More recently, in CPVT-patient induced pluripotent stem cells-derived cardiomyocytes, dantrolene restored normal calcium spark properties and rescued the arrhythmogenic phenotype.13,14 Further clinical trials are needed to confirm the safety and effectiveness in CPVT patients with RyR2 mutations. Left cardiac sympathetic denervation (LCSD) can be performed in patients who are not well controlled by drug therapy. Several case reports and a small case series have shown excellent results of LCSD in severely affected CPVT patients. Finally, implantable cardioverter defibrillators should be recommended for CPVT patients with syncope and/or documented sustained VT despite appropriate medications (Figure 2).
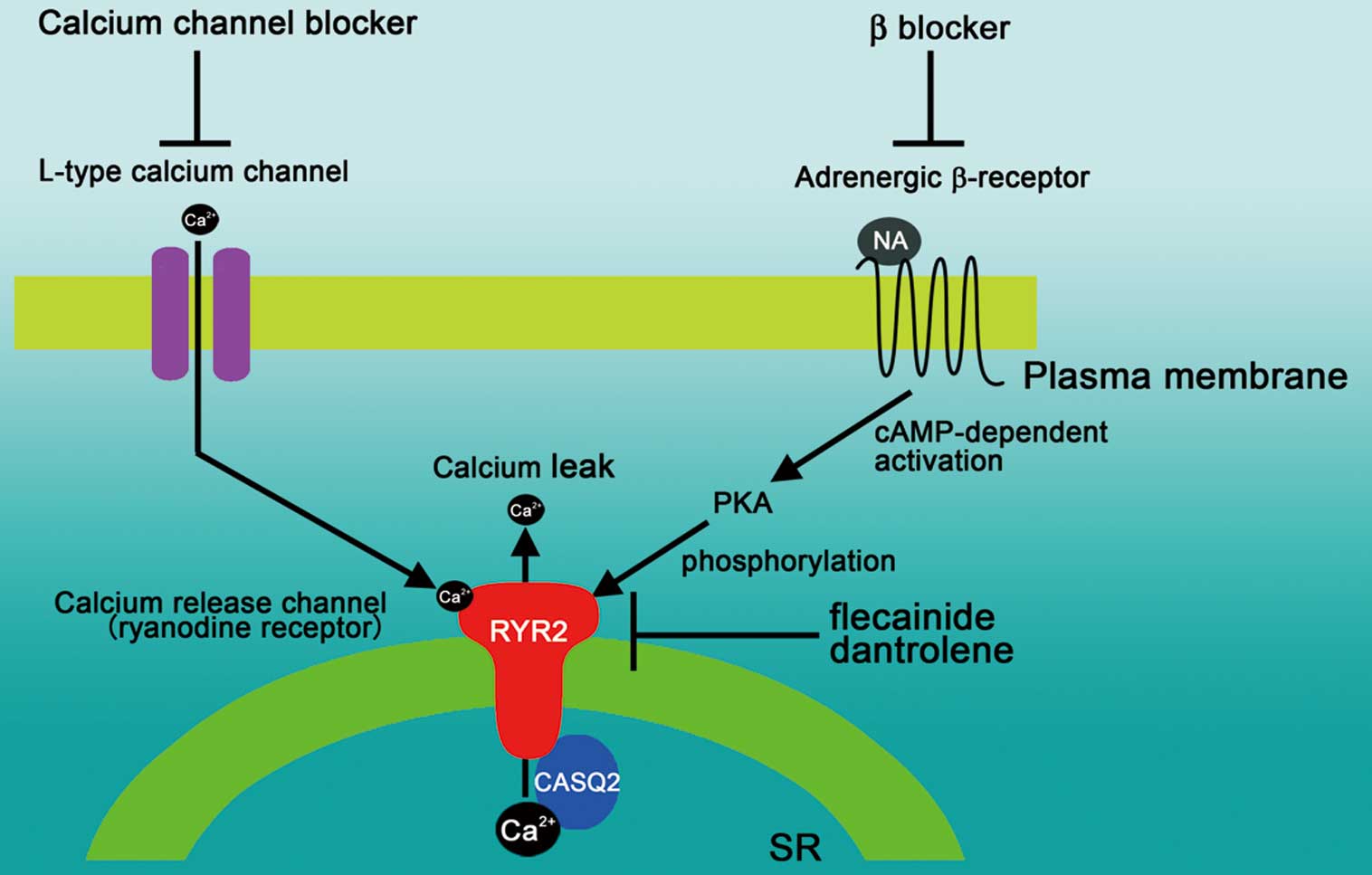
Molecular and signal targets in cardiac myocytes of established and potential medical treatments for catecholaminergic polymorphic ventricular tachycardia. CASQ2, calsequestrin; NA, noradrenaline; PKA, protein kinase A; RYR2, ryanodine receptor 2; SR, sarcoplasmic reticulum.
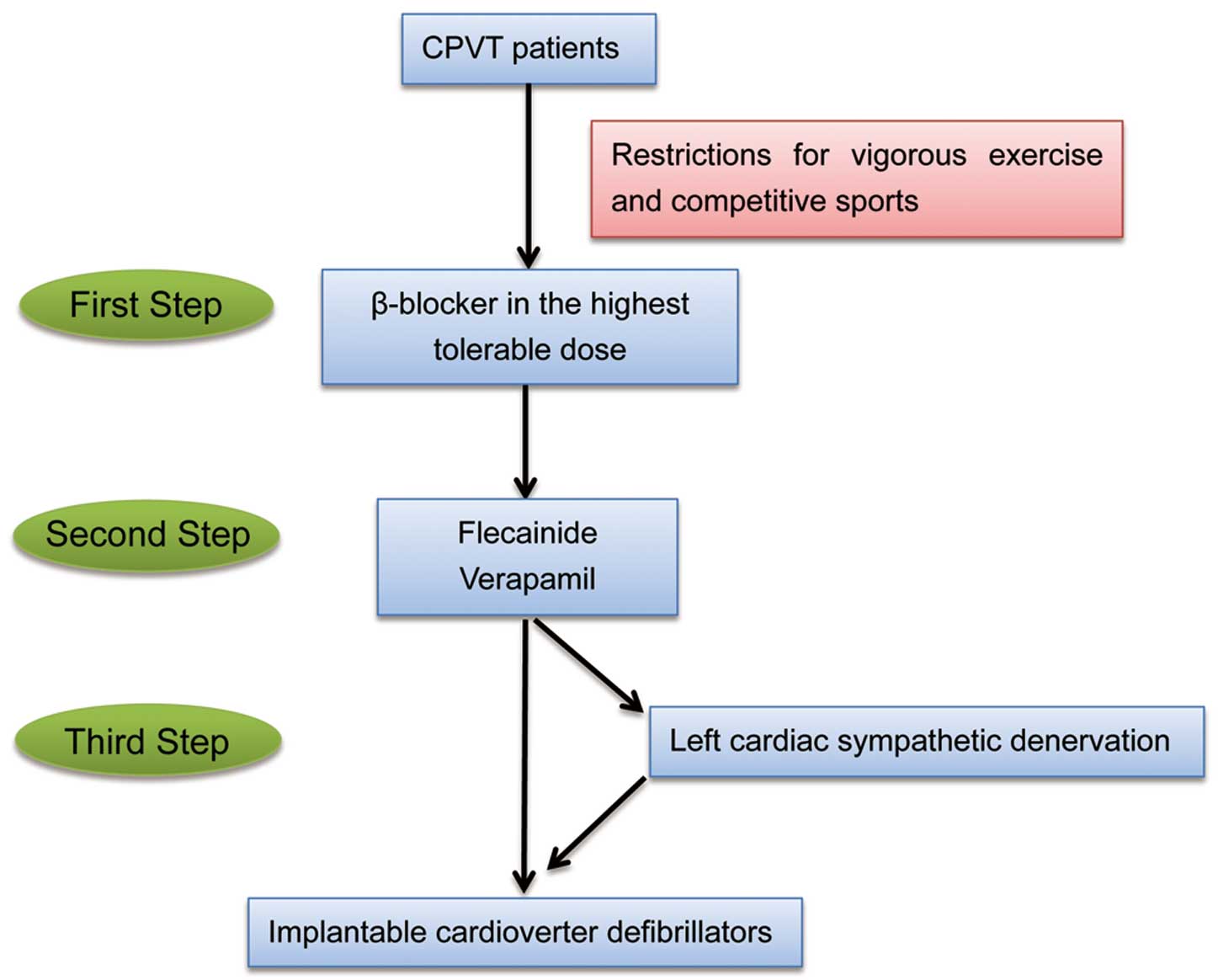
Flow chart of treatment for catecholaminergic polymorphic ventricular tachycardia.
Although the pathophysiological background and appropriate treatment are well understood, data for risk stratification are lacking,5 particularly for identifying those CPVT patients with a low risk of arrhythmic events in whom treatment can be withheld. Previous studies have shown that genotype-positive relatives demonstrate a wide variety of phenotypes. In a study of 116 drug-free relatives carrying the RYR2 mutation, 50% had a CPVT phenotype on initial cardiac examination, including 25% with non-sustained VT.15 Interestingly, mutation location may be associated with the severity of phenotype. Relatives with mutations in the C-terminal channel-forming domain of RYR2 had increased odds of non-sustained VT compared with those carrying mutations in the N terminal domain (odds ratio, 4.1; 95% confidence interval, 1.5–11.5).16 Similarly, exercise stress testing can be a useful tool for evaluation of asymptomatic relatives of CPVT probands. However, with the present condition, all phenotypically and/or genotypically diagnosed CPVT patients should receive active therapy. Larger cohorts, and multicenter CPVT patient registries are needed to elucidate the exact arrhythmic event rates in different subgroups, as well as the factors that identify individuals at high and low risk, especially in asymptomatic CPVT-associated mutation carriers. Furthermore, the role of genetic and environmental modifiers of the CPVT phenotype17 and the genetic background of current genotype-negative cases should be addressed in future studies.
None.
None declared.