Review
Development of Novel Cyclizations via Rhodacycle Intermediate and Its Application to Synthetic Organic Chemistry
2015 Volume 63 Issue 6 Pages 397-407
Details
2015 Volume 63 Issue 6 Pages 397-407
Novel Rh(I)-catalyzed cyclizations through a different type of rhodacycle intermediate which is formed by hydroacylation of 4,6-dienal or oxidative addition of diene and alkene are described. Hydroacylation of 4,6-dienal afforded various 7-membered rings in good to high yields, while cycloisomerization of diene and alkene provided 5- or 6-membered rings in good yields. On the basis of these studies, we have also succeeded in developing the sequential reaction of hydroacylation followed by cycloisomerization to produce bicyclic compounds in a stereoselective manner and thus this reaction was expanded to the synthesis of epiglobulol. Furthermore, both Rh(I)-catalyzed hydroacylation and cycloisomerization using ionic liquid (IL) as a solvent were investigated and it was found that the IL recovered after the reaction, which contains the Rh(I) catalyst, could be recycled several times without loss of catalytic activity.
Rh(I)-catalyzed intramolecular hydroacylation of C–C multiple bonds is an efficient and straightforward strategy for the construction of complex cyclic ketone derivatives from acyclic aldehyde in one-pot.1) In 1972, Sakai et al. were the first to report Wilkinson catalyst mediated hydroacylation of 4-alkenals to afford 5-membered carbocyclic ketones.2) After that, this hydroacylation of 4-alkenals has been expanded to a catalytic process3,4) and asymmetric reaction.5–18) The proposed reaction mechanism is shown in Chart 1. Initially, oxidative addition of the C–H bond of the aldehyde moiety to the rhodium complex occurs to form acylrhodium intermediate i followed by insertion of an alkene moiety to the Rh–H bond to give the 6-membered rhodacycle intermediate ii. Finally, reductive elimination from the rhodacycle intermediate ii occurs to give the 5-membered ketone.

As above-mentioned, there are many reports on the construction of 5-membered carbocyclic ring ketones through a hydroacylation process and its application to the synthesis of natural products.16,17) However, it is difficult to expand this hydroacylation to the construction of larger sized carbocyclic ring compounds because the side reactions are in competition with other reactions such as decarbonylation of the carbonyl group and isomerization of the alkene moiety in the substrate. In 1991, Gable and Benz reported that hydroacylation of 5-alkenals 3 afforded the 6-membered ring ketone 4 in moderate yield even though they used a 30 mol% Rh(I) complex19) (Chart 2, eq. 1). Tanaka and colleagues also found that hydoacylation of 6-alkynals 5 instead of 5-alkenal gave the cyclohexanone derivative 6 in high yield20) (Chart 2, eq. 2).

On the other hand, Fürstner and Aïssa reported the 7-membered ring 8 could be synthesized by hydroacylation of methylene cyclopropane 7.21,22) This reaction proceeded through hydrorhodation of the alkene moiety followed by C–C bond cleavage of the cyclopropane moiety (Chart 3, eq. 1). Shair and colleagues revealed that hydroacylation of vinyl cyclopropane 9 through rhodacycle intermediate iv provided 8-membered carbocyclic compound 10 in good yield23) (Chart 3, eq. 2). Furthermore, Aïssa and colleagues also found that the 8-membered carbocyclic ketone 12 was obtained through hydroacylation of methylene cyclobutane derivative 1124,25) (Chart 3, eq. 3). Although there are several reports on construction of larger sized carbocyclic compounds through hydroacylation, these have some limitations and a new reaction through hyadroacylation is therefore required. In this review, we present a comprehensive overview of our research on novel Rh(I)-catalyzed hydroacylation of 4,6-dienal to afford a 7-membered ring compound. We also describe a new cycloisomerization reaction of diene with alkene, which was found during investigation of the above-mentioned hydroacylation. On the basis of these studies, we have also succeeded in developing the sequential reaction of hydroacylation followed by cycloisomerization to produce a bicyclic compound from acyclic substrate in one-pot and thus this reaction was expanded to the synthesis of natural product. Furthermore, these two cyclizations carried out in ionic liquids (ILs) as reaction media with the aim of developing environmentally benign processes is also reported.

Initially, the hydroacylation of 4,6-dienals 13 was investigated in order to expand hydroacylation of the C–C multiple bond to construct larger sized carbocyclic ketones (Chart 4). That is, rhodacycle intermediate vi would be formed initially followed by isomerization to rhodacycle intermediate viii via a π-allylrhodium intermediate vii. If reductive elimination from viii occurs, the 7-membered cyclic ketone 14 would be formed.

Fortunately, it was found that the reaction of 13a with 10 mol% of [Rh(dppe)]ClO4 in 1,2-dichloroethane (DCE) at 65°C for 18 h afforded cycloheptenone 14a in 62% yield along with cyclopentanones 15a and 15a′ in 13% and 6% yields, respectively. It was thought that cycloheptenone 14a was produced through reductive elimination from rhodacycle intermediate viii, while reductive elimination from rhodacycle intermediate vi afforded 15a or 15a′ (Chart 5).

Next, the influence of the olefinic geometry of substrate 13a on reactivity toward this cyclization was carefully examined (Table 1). The cyclization of (4E,6E)-13a or (4Z,6E)-13a, both having an E-alkene at the C6 position, gave 7-membered ring compound 14a as the major product. On the other hand, cyclization of (4E,6Z)-1a, having a Z-alkene at the C6 position, gave 5-membered ring compound 15a as a major product instead of 7-membered ring compound 14a. These results indicate that the olefinic geometry of the diene moiety in 4,6-dienals has an influences on the reaction course.
 | |||||
|---|---|---|---|---|---|
| Substrate | Time (h) | 14a (%) | 15a (%) | 15a′ (%) | 13a rec. (%) |
| (4E,6E)-13a | 18 | 62 | 13a) | 6 | — |
| (4Z,6E)-13a | 16 | 65 | 11a) | 5 | — |
| (4E,6Z)-13a | 18 | Trace | 49b) | Trace | 38 |
a) E-Alkene. b) Z-Alkene.
Next, the cyclization of various 4,6-dienals was investigated under the similar conditions (Table 2). 4,6-Dienal 13b, having a cyclohexane moiety, afforded 7-membered ring 14b′ in 66% yields as a major product (run 1). The cyclization of 13c, having a benzyl ether in a tether, gave the mixture of 14c and 14c′ in good yield (run 2). On the other hand, when 13d and 13e, having a terminal diene moiety, were used as substrates, 5-membered rings (15d′ and 15e′) instead of 7-membered rings were obtained as a major product (runs 3 and 4).
| Run | Substrate | Time (h) | 7-Membered ring (14) | 5-Membered ring (15) |
|---|---|---|---|---|
| 1 |  | 24 |  |  |
| 2b) |  | 24 |  |  |
| 3c) |  | 17 |  |  |
| 4 |  | 16 | Not obtained |  |
a) All reactions were carried out in the presence of [Rh(dppe)]ClO4 (10 mol%) in DCE at 65°C. b) Reaction temperature was 45°C. c) The yields were determined by GC. d) The cyclized products were obtained as a single isomer although the stereochemistry has not been determined.
The hydroacylation of 4,6-dienals was expanded to the construction of a bicyclic compound having a 7-membered ring (Chart 6). The cyclization of 13f under similar conditions gave the bicyclic compound having 7-membered rings 14f and 14f′ as a major product, while the cyclization of 13g, having no substituent at the terminus of the diene moiety, afforded the bicyclic compound having 5-membered rings 15g′ as a major product.

These results suggest that there are two important points in order to obtain the 7-membered ring as a major product (Chart 7): 1) 4,6-Dienal 13 needs to have an E-alkene at the C6 position. 2) 4,6-Dienal 13 needs to have an alkyl substituent at the C7 position.
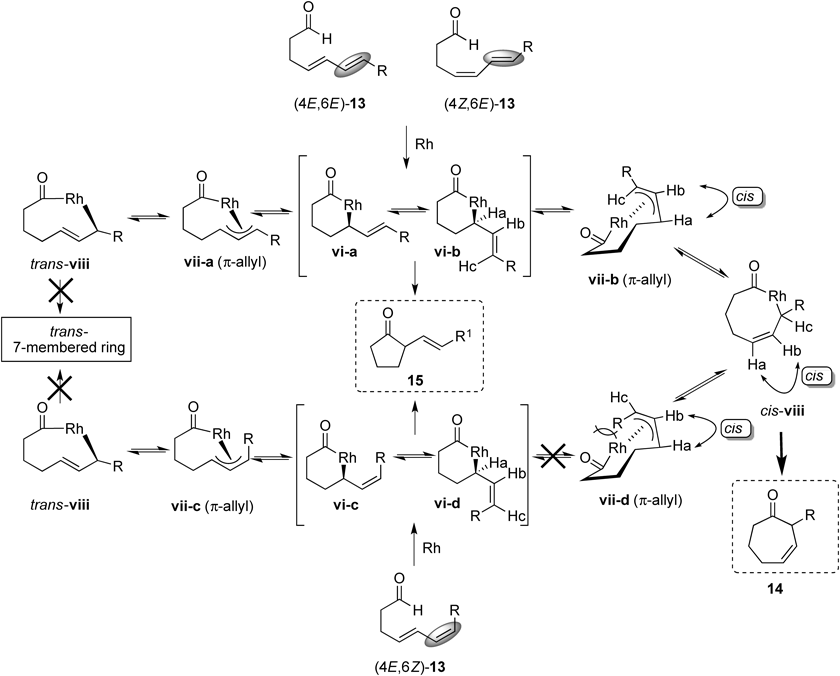
A plausible reaction mechanism is shown in Chart 8. The reaction of (4E,6E)-13 or (4Z,6E)-13 with a Rh(I) catalyst gives rhodacycle intermediates vi-a and vi-b, which might be in equilibrium with π-allylrhodium intermediates vii-a and vii-b. 7-Membered ring 14 should be produced through reductive elimination from rhodacycle intermediate cis-viii, while 5-membered ring 15 should be formed through reductive elimination from vi-a or vi-b. In the cyclization of (4E,6E)-13 and (4Z,6E)-13 having an E-alkene at the C6 position, 7-membered ring 14 was produced in preference to cyclopentanone 15. These results indicate that the equilibrium among the intermediates vi-a, vi-b, vii-a, vii-b, and cis-viii would lie to cis-viii although the reason is not clear. On the other hand, in the cyclization of (4E,6Z)-13 having an Z-alkene at the C6 position, rhodacycle intermediates vi-c and vi-d should be produced, which might be also in equilibrium with π-allylrhodium intermediates vii-c and vii-d. It was thought that the π-allylrhodium intermediate vii-d was unstable due to steric repulsion between the R-substituent at the C7 position and the rhodium metal center in the complex. Therefore, 5-membered ring 15 would be produced as a major product in the cyclization of (4E,6Z)-13. In all cases, the 8-membered rhodacycle intermediate trans-viii might be formed via π-allylrhodium intermediate vii-a or vii-c. However, it is apparent that a trans-seven-membered ring compound cannot be produced due to its lability. Furthermore, the alkyl substituent (R) at the C7 position in the substrate is needed in order to obtain 7-membered ring 14 as a major product. It is thought that a substituent plays an important role as a donating group to stabilize the buildup of positive charge on the terminal carbon of the diene moiety in cis-viii, which might promote the formation of 7-membered ring 14.
During the investigation of hydroacylation of 4,6-dienals, it was found that cyclization of 4,6-dienal 16a, having an olefin in the tether, gave 5-membered cyclic compound 17a instead of 7-membered ring 14h in high yield (Chart 9). This result suggests that cycloisomerization between 1,3-diene and alkene30–34) instead of hydroacylation of 4,6-dienal occurs to produce 5-membered cyclic compound 17a.

Although there are several examples of Rh(I)-catalyzed [4+2] cycloaddition of diene with alkene,35–42) cycloisomerization between a diene and alkene catalyzed by Rh(I) complex is still not available in the literature. Thus, this cycloisomerization of various substrates was examined (Table 3). Initially, the cyclization of (2E)-16b having no aldehyde moiety was carried out under the same conditions. As a result, it was found that 5-membered cyclic compound 17b was obtained in 85% yield (run 1). On the other hand, the reaction of (2Z)-16b, an olefinic isomer of (2E)-16b with respect to C2 carbon, gave 17b in 80% yield (run 2). Triene 16c or 16d, having a terminal 1,3-diene or internal alkene, afforded the corresponding cyclic compound 17c or 17d in 59% or 77% yield, respectively (runs 3 and 4). The substrate 16e, having a silyloxy group, was applicable for this reaction and 5-membered ring 17e was obtained in 85% yield (run 5). Furthermore, it was found that this cycloisomerization was expanded to the construction of 6-membered ring 17f (run 6).
| Run | Substrate | Time (h) | Product |
|---|---|---|---|
| 1 |  | 2 |  |
| 2 |  | 14 |  |
| 3 |  | 12 |  |
| 4 |  | 18 |  |
| 5 |  | 4 |  |
| 6 |  | 18 |  |
a) All reactions were carried out in DCE using 10 mol% [Rh(dppe)]ClO4 complex generated in situ from [Rh(nbd)(dppe)]ClO4 under an atmosphere of hydrogen at 65°C. R=CH2OBn.
Next, the construction of a heterocyclic 5-membered ring using this cycloisomerization was tested (Chart 10). Interestingly, it was found that a [4+2] cycloaddition reaction instead of this cycloisomerization proceeded in the case of 16g or 16h, having an oxygen or nitrogen atom between a 1,3-diene moiety and alkene, giving 18g or 18h in 37% or 92% yield, respectively (Chart 10).

A plausible reaction mechanism is depicted in Chart 11. Initially, 5-membered rhodacycle ix or 7-membered rhodacycle xii would be formed via oxidative cycloaddition of 1,3-diene and alkene in substrate 16 to a Rh(I) complex. 5-Membered rhodacycle ix would be in equilibrium with 7-membered rhodacycle xii via π-ally rhodium intermediate xi, and β-elimination of ix or xii would give rhodium hydride complex x or xiii. In the case of 16 (X=CR2) having no hetero atom between 1,3-diene and alkene, cycloisomerization product 17 would be formed through reductive elimination from x or xiii followed by an olefin isomerization reaction. On the other hand, in the case of 16 (X=O or NTs) having a hetero atom between 1,3-diene and alkene, [4+2] cycloaddition product 18 would be formed via reductive elimination from xii.

As mentioned above, we have succeeded in the development of Rh(I)-catalyzed hydroacylation of 4,6-dienel to afford 7-membered ring 14 and cycloisomerization between 1,3-diene with alkene to give 5-membered ring 17. These reactions were catalyzed by the same cationic Rh catalyst and proceeded under almost the same reaction conditions. We therefore planned to develop a new cascade reaction by combination of these reactions (Chart 12). That is, reaction of 4,6-dienal 19 having a 1,3-diene moiety with Rh complex would afford 7-membered ring 20 via hydroacylation, and then cycloisomerization of 1,3-diene with olefin of the resultant product 20 would occur to produce a bicyclo[5.3.0]decenone 21 in one-pot.

First of all, the cyclization of 19a was carried out in the presence of 10 mol% [Rh(dppe)]ClO4 in dichloroethane at 65°C. As a result, it was found that 7-membered ketone 20a was obtained in 66% yield instead of the desired sequential product 21a (Chart 13, eq. 1). Furthermore, when 7-membered ketone 20a was reacted with Rh(I) complex again at higher temperature, only a complex mixture not including sequential reaction product 21a was obtained (Chart 13, eq. 2).

These results indicate that cycloisomerization from hydroacylation product 20a to afford sequential product 21a catalyzed by Rh(I) complex in the second step does not proceed easily. In order to promote the second cycloisomerization step, we took advantage of the Thorpe–Ingold effect. That is, substrates 19b and 19c, which have a quaternary carbon center in a tether, were carefully examined (Chart 14). The sequential reaction of 19b proceeded smoothly, giving the desired bicyclic compound 21b in 44% yield in a stereoselective manner. On the other hand, when 19c was used as a substrate, the starting material and its olefinic isomers were recovered in 44% yield.

A possible reaction mechanism for the formation of 21b is shown in Chart 15. Initially oxidative addition of the C–H bond of the aldehyde moiety in substrate 19b followed by insertion of an alkene moiety occurs to give the 6-membered rhodacycle intermediate xv, which is isomerized to 8-membered rhodacycle intermediate xvii through π-allyl intermediate xvi. Reductive elimination from xvii takes place to give 7-membered ring 20b along with regeneration of Rh(I) complex. Next, oxidative addition of the diene and alkene moiety in 20b occurs stereoselectively to produce rhodacycle intermediate xviii followed by β-elimination from xviii to give rhodium hydride intermediate xix. Finally, reductive elimination from xix gives the bicyclic compound 21b along with regeneration of Rh(I) complex.

Encouraged by these results, we therefore planned for the synthesis of (±)-epiglobulol44–46) using bicyclic compound 21b as a key intermediate (Chart 16). Ketalization of 21b followed by hydrolysis of esters and decarboxylation gave mono carboxylic acid 22. Decarboxylation of 2247) afforded 23 followed by regioselective dibromocyclopropanation of internal olefin in 23 gave 24 in 31% yield along with starting material 23 in 56% yield.48) Introduction of two methyl groups into the cyclopropane moiety using a copper reagent gave 25 in 99% yield. An allyl moiety of cyclic compound 25 was converted to a methyl group using an ozonolysis of olefin in 25 followed by decarbonylation in 66% yield. Deprotection of the ketal 26 afforded (±)-apoaromadendrone (27), whose spectral data were completely identical with those reported in the literature.45) Finally, (±)-apoaromadendrone (27) was converted into (±)-epiglobulol (28) by use of MeLi according to a method described in the literature45) (Chart 16).

Reagents and conditions: (a) TMSOTf, TMSO(CH2)2OTMS, CH2Cl2, −78°C, quant.; (b) NaI, NaHCO3, DMF, 150°C; (c) LiOH·H2O, MeOH–H2O, 50°C, 2 steps 99%; (d) (EtO)2P(O)Cl, Et3N, MS 4A, THF, rt; (e) 2-Mercaptopyridine N-oxide sodium salt, tBuSH, toluene, reflux, 2 steps 65%; (f) BnEt3NCl, CHBr3, 50% NaOH aq., CH2Cl2, 0°C, 71% (conversion yield); (g) Me3CuLi2, Et2O, then MeI, rt, 99%; (h) O3, CH2Cl2, −78°C, then PPh3, rt; (i) RhCl(PPh3)3, PhCN, 140°C, 2 steps 66%; (j) FeCl3·SiO2, acetone, rt, 67%; (k) MeLi, Et2O, −78°C, 84%.
Guanacastepenes have attracted the interest of synthetic chemists around the world due to their unique structural features, which include two ring-junction quaternary methyl groups and a 5-7-6 tricyclic carbon skeleton50–53) (Chart 17). This family also shows excellent activity against methicillin-resistant Staphylococcus aureas (MRSA) and vancomycin-resistant Enterococcus faecalis (VREF) pathogens.54) Thus, we planned for the construction of tricyclic core skeleton using the above-mentioned Rh(I)-catalyzed sequential reaction of hydroacylation followed by cycloisomerization (Chart 17). That is, if substrate 29 having a cyclohexane ring is treated with Rh(I) complex, a 5-7-6 tricyclic compound 31, whose structure is similar to the core structure of guanacastepene family, would be produced through sequential reaction in one-pot.

Initially, the influence of the relative stereochemistry on the cyclohexane ring was carefully examined (Chart 18). The cyclization of syn-29 with 10 mol% [Rh(dppe)]ClO4 proceeded smoothly, giving the desired tricyclic syn-31 along with syn-32 which was produced through only hydroacylation. And in this reaction, anti-31 and anti-32 also were obtained in 7% and 23% yield, respectively, indicating that an isomerization with respect to the stereochemistry on the cyclohexane ring occurred during the sequential reaction. On the other hand, when anti-29 was used as a substrate, hydroacylation product anti-32 was obtained in 32% yield as a major product along with the desired tricyclic compound anti-31 in 9% yield. These results indicate that in order to obtain the desired tricyclic compound 31 as a major product, isomerization of syn-29 to anti-29 should be suppressed.

Thus, the effect of additives on this reaction to prevent isomerization from syn-29 to anti-29 was investigated (Table 4). When the cyclization of syn-29 was carried out in the presence of CaCO3, syn-31 was obtained in 50% yield along with syn-32 in 9% yield although a partial isomerization still occurred (run 2). On the other hand, the cyclization of syn-29 in the presence of MS 4A afforded the desired tricyclic compound syn-31 in 61% yield along with syn-32 in 12% yield (run 3). In this reaction, it was found that isomerization from syn-29 to anti-29 was suppressed completely, even though the reason was not still unclear.
 | ||||||
|---|---|---|---|---|---|---|
| Run | Additive | syn : anti | Yields (%) | |||
| syn-31 | syn-32 | anti-31 | anti-32 | |||
| 1 | — | 58 : 42 | 34 | 8 | 7 | 23 |
| 2 | CaCO3b) | 80 : 20 | 50 | 9 | 6 | 9 |
| 3 | MS 4A | 100 : 0 | 61 (54)c) | 12 | — | — |
a) Yields were determined by GC analysis. b) CaCO3 was used 1.5 eq to syn-5. c) Isolated yield.
Ionic liquids (ILs) have attracted attention as reaction media in synthetic organic chemistry due to their unique properties (e.g. wide liquid range with a melting point around room temperature, low vapor pressure, high polarity, and thermal stability etc.).56–63) Furthermore, ILs are known to dissolve charged species such as organometallic compounds more readily than non-charged species, including most organic compounds, via ionic interactions. This property of ILs might enable easy separation of the catalyst and organic compounds by simple phase separation and recycling of the catalyst remaining in ILs. Thus, we examined Rh(I)-catalyzed two cyclizations using ILs as solvents with the aim of developing environmentally benign chemical processes.
Initially, the hydroacylation of 4-alkenal 33a was investigated using various ionic liquids as solvent (Table 5). In reactions using various ILs 35 having butylmethylimidazolium moiety as a cation [BMI]+ (BMI=1-butyl-3-methyl imidazolium), the hydroacylation of 33a proceeded to give cyclopentanone derivative 34a in high yields (runs 1–3). On the other hand, the use of ILs 36 having a butyldimethylimidazolium moiety as a cation [BDMI]+ (BDMI=1-butyl-2,3-dimethyl imidazolium) also showed high yields even though the completion of the reaction needed a longer time than that in ILs 35 (runs 4–6).
 | |||
|---|---|---|---|
| Run | IL | Time (h) | Yield (%) |
| 1 | [BMI][BF4] (35a) | 20 | 95 |
| 2 | [BMI][PF6] (35b) | 144 | 90b) |
| 3 | [BMI][NTf2] (35c) | 48 | >99 |
| 4 | [BDMI][BF4] (36a) | 168 | >99b) |
| 5 | [BDMI][PF6] (36b) | 144 | 93b) |
| 6 | [BDMI][NTf2] (36c) | 120 | >99b) |
a) All reactions (except for run 1) were carried out in a mixed solvent of DCE and IL (ratio of 1.0 : 3.2). b) GC yield.
Next the cyclization of various substrates was examined in [BMI][NTf2] (NTf2=bis(trifluoromethane sulfonyl) amide)as a solvent (Table 6). The reaction of 33b, having a phenyl group at the C3 position, and 33c, 1,1-disubstituted alkene, gave the cyclopentanones 34a and 34c in 94% and 91% yields, respectively (runs 1 and 2). 33d and 33e, having a conjugated alkene, afforded the desired products 34d and 34e in quantitative yields, respectively. Furthermore, when 4,6-dienal 13a was used as a substrate, 7-membered cyclic compound 14a was obtained in 68% yield after 3 h. It was found that this reaction was greatly accelerated by the use of IL as a reaction solvent (Chart 5 vs. Table 6, run 5).
| Run | Substrate | Time (h) | Product | Yield (%) |
|---|---|---|---|---|
| 1 |  | 8 |  | 94 |
| 2 |  | 8 |  | 91b) |
 |  | |||
| 3 | 33d (R=Me) | 1 | 34d | >99 |
| 4 | 33e (R=Ph) | 1 | 34e | >99 |
| 5 |  | 3 |  | 68 |
a) All reactions were carried out in the presence of 10 mol% [Rh(dppe)]ClO4 in a mixed solvent of [BMI][NTf2] and DCE (ratio of 3.2 : 1.0) at room temperature. b) cis : trans=43 : 57.
Finally we turned our attention to recycling of the catalyst remaining in the IL (Table 7). After the reaction of 33d had been completed, the product 34d was extracted from the reaction media with Et2O, and the remaining [BMI][NTf2] phase containing the cationic Rh(I) complex was dried under reduced pressure and reused without further treatment. In the reaction using [Rh(dppe)]ClO4 as a catalyst, the reaction could be repeated 10 times without significant loss of catalytic activity.
 | ||
|---|---|---|
| Rh Catalyst (mol%) | Cycle | Yield (%) |
| [Rh(dppe)]ClO4 (10) | 1 | >99 |
| 2 | 97 | |
| 3 | >99 | |
| 4 | 99 | |
| 5 | 98 | |
| 6 | 99 | |
| 7 | >99 | |
| 8 | >99 | |
| 9 | 98 | |
| 10 | >99 | |
a) All reactions were carried out in [BMI][NTf2].
As mentioned above, we succeeded in the recycling of the Rh(I) complex in hydroacylation using IL as a solvent. Next, the cycloisomerization catalyzed by the same Rh(I) complex was also examined using IL as a solvent (Table 8). As a result, it was found that the reaction in [BMI][NTf2] showed high yield and the completion of the reaction needed only 2 h.
 | |||
|---|---|---|---|
| Run | Solvent | Time (h) | Yield (%) |
| 1 | [BMI][BF4] | 3 | 96 |
| 2 | [BMI][PF6] | 120 | 80 |
| 3 | [BMI][NTf2] | 2 | 98 |
| 4 | [BDMI][BF4] | 45 | 96 |
| 5 | [BDMI][PF6] | 72 | —a) |
| 6 | [BDMI][NTf2] | 4 | 88 |
a) Complex mixture.
Next, the recycling of the IL containing the cationic Rh(I) complex was tested (Table 9). It was found that the reaction using [BMI][NTf2] as a solvent, the reaction could be repeated only 3 times. This result indicated that the recyclability of the catalyst in this cycloisomerization was less than that in the above-mentioned hydroacylation even though these reactions were subjected to almost the same conditions, with the exception of the reaction temperature. The exchange of the counter anion of [Rh(dppe)]ClO4 from ClO4− into NTf2− of [BMI][NTf2] occurred to generate [Rh(dppe)]NTf2, which caused a reduction in the number of recyclings of the catalyst. To confirm this hypothesis, the cyclization of 16b using 10 mol% [Rh(dppe)]NTf2 in [BMI][NTf2] was carried out under the same conditions. Surprisingly, it was found that the reaction proceeded smoothly, giving the cyclic compound 17b in 96% yield and the IL containing Rh(I) complex could be recycled 4 times. Next, cycloisomerization of 16b using [BDMI][NTf2] instead of [BMI][NTf2], having a methyl group on the C-2 position, was also investigated. Interestingly, in the case of the reaction in [BDMI][NTf2], the IL containing the cationic Rh(I) catalyst could be recycled 8 times without the significant loss of any catalytic activity.
 | ||||
|---|---|---|---|---|
| Rh Catalyst | IL | Cycle | Time (h) | Yield (%) |
| [Rh(dppe)]ClO4 | [BMI][NTf2] | 1 | 3 | 95 |
| 2 | 3 | 92 | ||
| 3 | 5 | 93 | ||
| 4 | 48 | 44a) | ||
| [Rh(dppe)]NTf2 | [BMI][NTf2] | 1 | 2 | 96 |
| 2 | 2 | 95 | ||
| 3 | 2 | 94 | ||
| 4 | 2 | 91 | ||
| 5 | 6 | 77a) | ||
| [Rh(dppe)]NTf2 | [BDMI][NTf2] | 1 | 4 | 99 |
| 2 | 4 | 98 | ||
| 3 | 4 | 97 | ||
| 4 | 4 | 96 | ||
| 5 | 4 | 95 | ||
| 6 | 4 | 96 | ||
| 7 | 4 | 94 | ||
| 8 | 5 | 82 | ||
a) Complex mixture.
In order to reduce the amount of Rh(I) catalyst, various additives in the cycloisomerization of 16b were investigated (Chart 19). It was found that the use of 2,2,2-trifluoroethanol (TFE) as an additive enabled the catalyst loading to be reduced to 5 mol% and the IL containing Rh(I) complex to be reused 7 times without significant loss of catalytic activity.

Encouraged by these results, Rh(I)-catalyzed cycloisomerization of various substrates in [BDMI][NTf2] was investigated (Chart 20). In all cases using [BDMI][NTf2], the cycloisomerization proceeded smoothly to give the desired compounds 17c, d and f in higher yields than those in dichloroethane (Table 3), and the catalyst could be reused at least 3 times without significant loss of catalytic activity.

We have succeeded in the development of novel Rh(I)-catalyzed hydroacylation of 4,6-dienals to afford 7-membered rings and cycloisomerization between 1,3-diene and alkene to give 5- or 6-membered rings in good to high yields, respectively. On the basis of these studies, sequential reaction of hydroacylation of 4,6-dienals followed by cycloisomerization between 1,3-diene and alkene to give a bicyclo[5.3.0]decenone in one-pot in a stereoselective manner was also developed and the synthesis of natural product was accomplished by the use of the sequential reaction. Furthermore, we found that Rh(I)-catalyzed these cyclizations were carried out in IL as a reaction media and the IL containing Rh(I) catalyst could be used several times without significant loss of catalytic activity. We believe that these studies will further highlight the usefulness of this methodology in synthetic organic chemistry.
All of the research in this review was conducted at the Faculty of Pharmaceutical Sciences, Hokkaido University. The author is grateful to Professor Yoshihiro Sato for his continued support and helpful discussions. The author would like to express his sincere appreciation to all of the students whose names are acknowledged in our publications cited in this review. These studies were supported in part by a Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science, and Research Grant from the Akiyama Foundation.
The author declares no conflict of interest.