Notes
Synthesis of a Model Compound of the Unique Meroterpenoids Cryptolaevilactones
2022 年 70 巻 10 号 p. 740-747
詳細
2022 年 70 巻 10 号 p. 740-747
Cryptolaevilactones (CLs) A–L, found in the leaves and twigs of Cryptocarya laevigata, are unique natural meroterpenoids with a spiro[3.5]nonane skeleton. We report the total synthesis of a simplified model compound of CLs A–C. The synthetic route included introduction of a styryl group and coupling of a lactone moiety to a spiro ring system, which was constructed by a pinacol-like rearrangement.
Twelve meroterpenoid lactones [cryptolaevilactones (CLs) A–L (1–12)] were first isolated from a 50% MeOH/CH2Cl2 extract (N025439) of the leaves and twigs of Cryptocarya laevigata by our group1,2) (Fig. 1). The compounds’ unique structural core is a spiro[3.5]nonane skeleton, which might be biosynthesized through a [2 + 2] cycloaddition of the monoterpene β-phellandrene with a polyketide produced from cinnamoyl-CoA and five units of malonyl-CoA. CLs A–C and L (1–4) are diastereomers due to different configurations at six chiral centers, C-6, C-8, C-11, C-12, C-1′, and C-4′. Compounds 5–12 are likely derived from CLs A–C or L by intramolecular Michael additions of the lactone moiety to form bicyclic lactones D–F (5–7), by dehydration to produce analogous Δ8,9 CLs G–J (8–11), or by acetylation of the hydroxy group on C-8 of 2 to produce CL K (12). The absolute configurations at C-8 of CLs A–C (1–3) were determined by using a modified Mosher method.1) Subsequently, nuclear Overhauser effect spectroscopy (NOESY) correlations between H-6 and H-8 of the related Michael adducts, CLs D–F (5–7), revealed that these hydrogen atoms have a syn relationship. The relative configurations of the four asymmetric carbons on the spiro ring were also assigned from NOESY correlations and ECD calculations.2) These structurally unique natural products were isolated in only limited amounts and their biological properties of these compounds could not be evaluated; thus, a total synthesis was warranted.
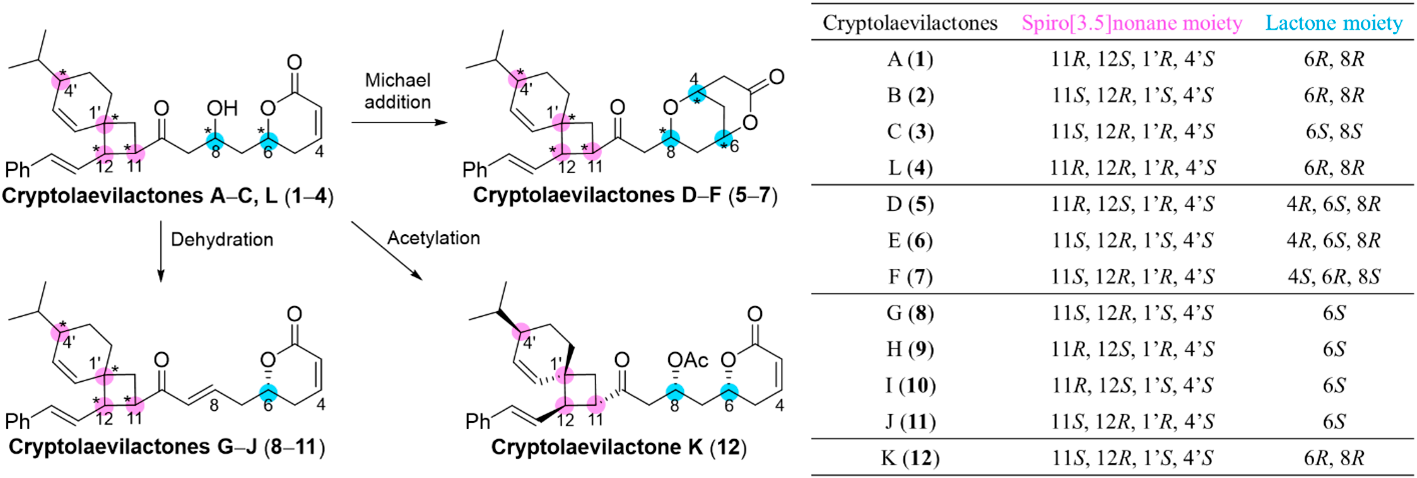
The synthesis of CLs A–C would enable the comprehensive preparation of all CLs. For this purpose, we first designed a simplified model compound 13 (Chart 1), which has two fewer asymmetric carbons on the spiro ring than CLs A–C, to avoid the complex mixtures resulting from uncontrollable chiral centers.

Model compound 13 was divided into three main fragments, the styrene, spiro-nonane, and lactone-chain moieties (Chart 1). In our proposed synthetic strategies, the three fragments would be synthesized independently and connected via different routes. The introductions of the styrene and lactone moieties onto the spiro-nonane skeleton were expected to be key, but difficult, steps. Two retrosynthetic routes A and B were examined (Chart 1). Both routes employ the common intermediate 7,7-dimethylspiro[3.5]nonan-1-one (14), obtained via a pinacol-like rearrangement of the tertiary alcohol 15 produced from readily available 4,4-dimethylcyclohexanone (16).3) The two routes differ primarily by using a one-carbon homologation (route A) or two-carbon homologation (route B) of 14 prior to connection of the lactone-chain. In route A, the final synthetic step is lactone construction in precursor 17, obtained through a Corey–Seebach reaction between the known epoxide 184–6) and the functionalized spiro-nonane 19. The latter compound would be produced from 14 via a one-carbon homologation to add 1,3-dithiane to 14 forming 2-(1,3-dithian-2-yl)-7,7-dimethylspiro[3.5]nonan-1-one (20) followed by conversion of the oxo group in 20 to the styrene moiety in 19. In route B, the final target 13 would be prepared by an aldol reaction between the known (R)-2-(6-oxo-3,6-dihydro-2H-pyran-2-yl)acetaldehyde (22)7) and the synthesized acetyl spiro-nonane 23, obtained by a two-carbon homologation of 14 to give 24 followed by construction of the styrene moiety.
The required common intermediate 14 was synthesized by using Trost’s and Undheim’s methods8–10) (Chart 2). The treatment of cyclohexanone 163) with cyclopropyl phenyl sulfide provided tertiary alcohol 15 in 87% yield. The pinacol-like rearrangement of 15 with an aqueous solution of HBF4 as an acid catalyst successfully generated spiro ketone 14, which was converted to the related silyl enol ether 26 prior to insertion of the appropriate functional group.

Route A required the introduction of a thioketal group at the α-position of ketone 14 (Chart 3). Using Paterson’s method,11) 1,3-dithienium fluoroborate was added to silyl enol ether 26 to successfully produce thioketal 20. Various attempts at the insertion of the styrene moiety, including the use of (styryl)lithium or halogenation of the ketone group, did not yield the desired compound. However, a Van Leusen reaction of ketone 20 using toluenesulfonylmethyl isocyanide (TosMIC) successfully produced the homologous nitrile 27 in 58% yield with only trans-oriented thioketal and nitrile moieties. Then, nitrile 27 was treated with benzyl magnesium chloride to provide ketone 28, which was reduced with NaBH4 to obtain alcohol 29 in excellent yield. Dehydration of the secondary alcohol in 29 was accomplished by treatment with mesyl chloride followed by t-BuOK to selectively give the trans-alkene in the styryl-substituted product 19. After the successful 5-step introduction of the styryl group, we attempted the Corey–Seebach reaction between 19 and epoxide 18.4–6) However, the dithianide anion generated by n-BuLi did not directly attack the epoxide in 18 to produce the target compound 17. Instead, unwanted compound 30 was predominantly generated; a possible mechanism would involve ring opening of the cyclobutane followed by an attack of the resulting benzylic anion on the epoxide.
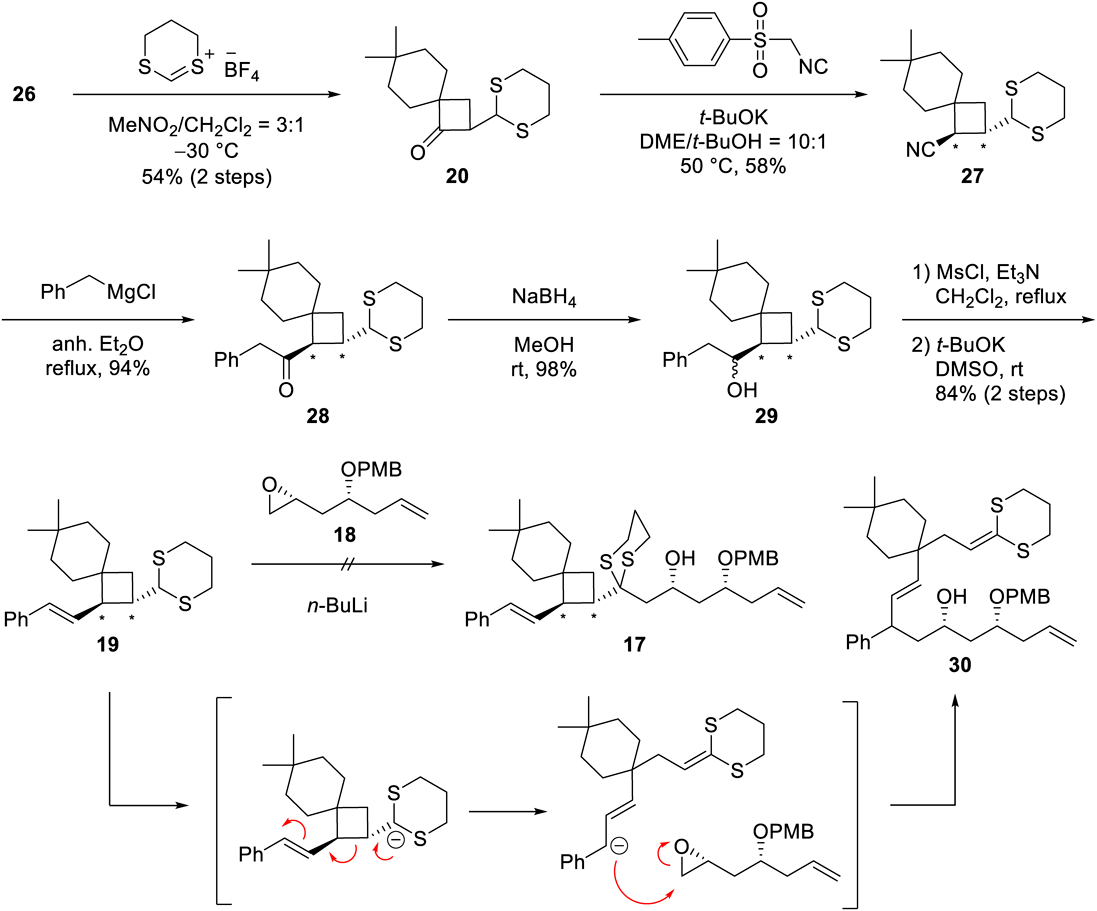
After this disappointing result, we switched our synthetic efforts to route B (Chart 4), which connects the lactone and spiro moieties at a different point from that in route A. The preparations of the important acetylated intermediate 23 and its silyl enol ether 35 began with the treatment of 26, the silyl enol ether of common intermediate 14, with acetaldehyde in the presence of tetrabutylammonium fluoride (TBAF).12,13) The secondary alcohol 31 was obtained in moderate yield and protected as its methoxymethyl (MOM) ether (24). Subsequently, the styryl group was inserted using the same 5-step procedure described in route A to produce 34 with trans-oriented side chains on the cyclobutane; no cis-oriented product was obtained. The deprotection of the MOM group under an acidic condition followed by oxidation of the resulting alcohol with Dess–Martin periodinane (DMP) afforded acetyl spiro-nonane 23 in high yield. The NOESY correlations between H-9/H-12 and H-11/H-13 supported the trans-relationship of the acetyl and styryl groups on the cyclobutane in 23. We also prepared the related silyl enol ether 35, so that we could use a Mukaiyama reaction rather than a classic aldol reaction in the final coupling step.
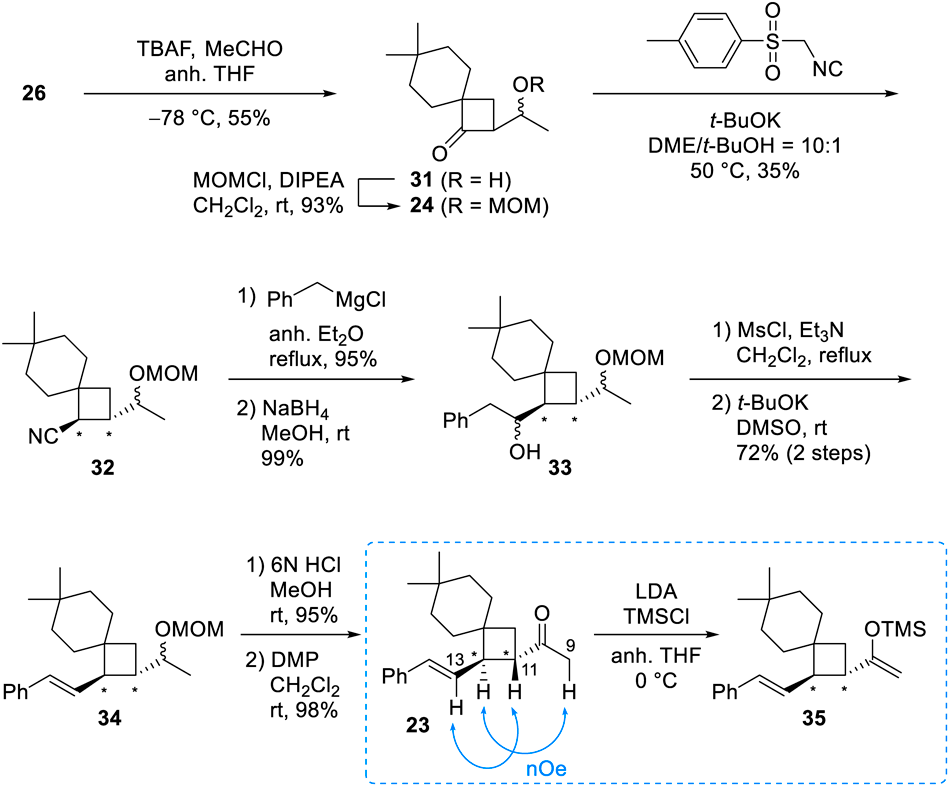
In the route B retrosynthetic design of our model compound of CLs, the final step was the coupling of acetyl spiro-nonane 23 and aldehyde 227) (Chart 1). Various reaction types and conditions were attempted, and the results are shown in Table 1. A Mukaiyama aldol reaction14,15) of 35, the silyl enol ether of 23, with 22 in the presence of SnCl4 at 0 °C gave a complex mixture (Entry 1), while only acetyl spiro-nonane 23 was recovered at a lower temperature (Entry 2). The use of BF3·OEt2 at 0 °C also gave only the parent ketone 23 without any reaction with aldehyde 22 (Entry 3). However, the same reaction at −50 °C generated the desired aldol adducts (13) in 19% yield as a mixture of four diastereomers (16 : 16 : 34 : 34 ratio), which were purified by chiral HPLC (CHIRAL ART Cellulose-SC, YMC). The stereochemistry of each aldol adduct was determined from an analysis of 1H-NMR data as described below. While unfortunately, the undesired C-6,8-anti compounds, anti-13a and 13b, were the main products, the desired C-6,8-syn aldol adducts, syn-13a and 13b, were obtained, albeit in minor amounts (Entry 4). In an effort to possibly increase the reaction selectivity and the amounts of the wanted syn aldol adducts, we also tried Keck’s method with Ti(OiPr)4 and (S)-1,1′-bi-2-naphthol (BINOL)16) (Entry 5) and a stereo-controlled aldol reaction utilizing (−)-B-chlorodiisopinocampheylborane [(−)-DIP-Cl] derived boron enolates17,18) (Entry 6). However, these attempts failed to produce coupled products, instead leading to recovery of ketone 23 or decomposition, respectively.
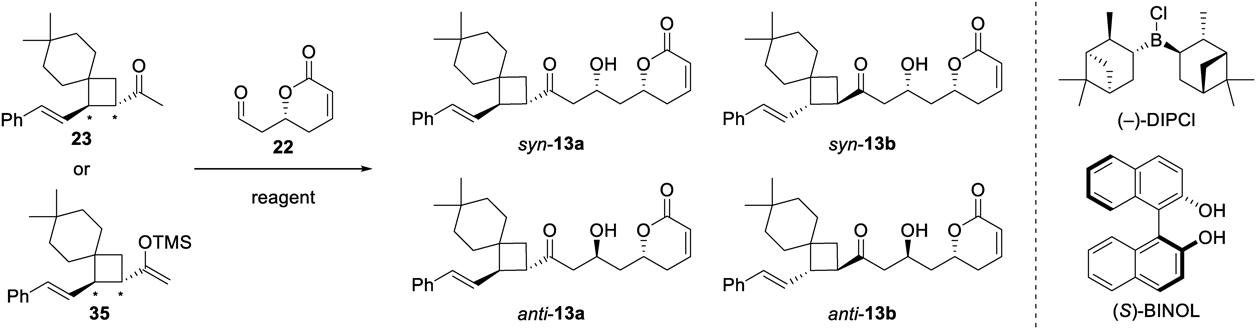 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | SMa) | Reagent (equiv.) | Solvent | Condition | Yield (%) | ||
| Temp. (°C) | Time (h) | (syn-13a/syn-13b/anti-13a/anti-13b) | 23 | ||||
| 1b) | 35 | SnCl4 (2.6) | CH2Cl2 | 0 | 1 | 0 | 0 |
| 2b) | 35 | SnCl4 (2.6) | CH2Cl2 | –50 | 3 | 0 | 75 |
| 3b) | 35 | BF3·OEt2 (2.5) | CH2Cl2 | 0 | 1 | 0 | 71 |
| 4b) | 35 | BF3·OEt2 (2.5) | CH2Cl2 | –50 | 3 | 19d) (16 : 16 : 34 : 34)e) | 60 |
| 5b) | 35 | Ti(OiPr)4 (1.6), (S)-BINOL (3.2) | THF | 0 to rt | 16 | 0 | 86 |
| 6c) | 23 | (−)-DIPCl (2.2), Et3N (3.1) | Et2O | –78 | 2 | 0 | 0 |
a) Starting material. b) The reactions were performed using 2.5 equiv. of 22. c) The reaction was performed using 5.6 equiv. of 22. d) Two step yields from 23. e) The ratio was determined from amounts of isolated products.
The identities of the C-6,8-syn 13 and C-6,8-anti 13 diastereomers were established through the comparison of the product 1H-NMR data with those of previously synthesized CL M2) and other natural products containing the same lactone portion, such as cryptolactones A1, A2, B1, and B2.19) As an example, Fig. 2 shows the 1H-NMR data comparison with CL M, which has C-6,8-syn oxygen functions. The syn or anti diastereomers of 13 could be clearly determined from the differences in the coupling patterns and chemical shifts of H-5 and H-7 in the 1H-NMR. Similar 1H-NMR differences were observed previously between cryptolactones A1 and B1, which have anti-stereochemistry, and cryptolactones A2 and B2, which have syn-stereochemistry.19)

In summary, we achieved the synthesis of a simplified model compound (13) of CLs A–C. Our synthetic route effectively introduced the styryl group onto the spiro[3.5]nonane core via a nitrile intermediate and subsequently added the lactone fragment in the final synthetic step to produce 13. By dividing the target compound into three fragments, our strategy offers the advantages of easy derivatization and ready application to asymmetric synthesis. Although the final aldol step must be improved, this synthetic route can certainly be applied to the first total synthesis of CLs.
NMR spectra were taken on a JEOL JNM-ECA600 or JNM-ECZ600R spectrometer and JNM-ECS400 spectrometer with tetramethylsilane (TMS) as an internal standard, and chemical shifts are expressed as δ values. High-resolution mass spectrometric data were obtained on a JEOL JMS-700 (FAB) or a JMS-T100TD (DART) mass spectrometer. Analytical and preparative TLC was carried out on precoated silica gel 60F254 and RP-18F254 plates (0.25- or 0.50-mm thickness; Merck, Germany). Column chromatography was performed using silica gel 60N (63–210 µm, Kanto Chemical). MPLC was performed with silica gel and C18 cartridges (Biotage, Uppsala, Sweden). Semi-preparative HPLC was conducted using CHIRAL ART Cellulose-SC (250 × 10.0 mmI.D.; YMC).
4,4-Dimethyl-1-[1-(phenylthio)cyclopropyl]cyclohexan-1-ol (15)To a stirred solution of cyclopropyl phenyl sulfide (0.17 mL, 1.20 mmol) in dry tetrahydrofuran (THF) (2.0 mL), n-BuLi (1.6 M in hexane, 0.74 mL, 1.18 mmol) was added dropwise at 0 °C and the mixture was stirred at this temperature for 2 h. A solution of 16 (98.9 mg, 0.785 mmol) in dry THF (0.8 mL) was added dropwise to the mixture, which was stirred at same temperature for another 2 h. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted three times with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residual material was purified by column chromatography on silica gel to give 15 (188.4 mg, 87%) as a colorless oil. 1H-NMR (600 MHz, CDCl3) δ: 7.40–7.45 (m, 2H), 7.22–7.28 (m, 2H), 7.11–7.15 (m, 1H), 1.67 (dt, J = 3.6, 14.4 Hz, 2H), 1.62 (s, 1H), 1.48–1.55 (m, 4H), 1.23 (dd, J = 5.4, 6.6 Hz, 2H), 1.17–1.22 (m, 2H), 0.94 (dd, J = 5.4, 6.4 Hz, 2H), 0.91 (s, 3H), 0.84 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 137.8, 128.6, 128.2, 125.4, 73.1, 35.8, 34.5, 32.6, 30.6, 29.5, 23.2, 13.5. High resolution (HR)MS-FAB (m/z): [M + H]+ Calcd for C17H25OS, 277.1626; Found, 277.1628.
7,7-Dimethylspiro[3.5]nonan-1-one (14)To a solution of 15 (1.29 g, 4.68 mmol) in dry Et2O (25.0 mL) was added aqueous 48% HBF4 (4.3 mL, 32.8 mmol) and the mixture was refluxed for 6 h. Et2O was added to the reaction mixture and the solution was washed three times with saturated aqueous NaHCO3 and once with water. The organic solution was dried over Na2SO4 and concentrated and the residual material was purified by column chromatography on silica gel to give 14 (640 mg, 82%) as a colorless solid. 1H-NMR (400 MHz, CDCl3) δ: 2.96 (t, J = 8.4 Hz, 2H), 1.81 (d, J = 8.4 Hz, 2H), 1.75 (ddd, J = 3.6, 10.0, 13.6 Hz, 2H), 1.51–1.59 (m, 2H), 1.37–1.45 (m, 2H), 1.24 (ddd, J = 3.6, 10.0, 13.6 Hz, 2H), 0.91 (s, 3H), 0.90 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 216.4, 65.5, 53.5,* 41.3, 35.1, 31.0,* 29.9,* 29.4, 28.1, 26.5,* 23.9.
*These signals might be derived from the minor conformation of cyclohexane.
2-(1-Hydroxyethyl)-7,7-dimethylspiro[3.5]nonan-1-one (31)To a stirred solution of 14 (131.3 mg, 0.791 mmol) in CH2Cl2 (3.0 mL) were added Et3N (0.27 mL, 1.95 mmol) and TBSOTf (0.22 mL, 0.958 mmol) at 0 °C and the mixture was stirred at the same temperature for 2 h. The mixture was quenched with saturated aqueous NaHCO3 and extracted three times with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to give crude silyl enol ether 26, which was used in the next step without further purification. TBAF (1.0 M in THF, 0.95 mL, 0.950 mmol) was added to a suspension of 4 Å molecular sieves (300 mg) in dry THF (1.0 mL), stirred at room temperature (r.t.) for 2 h, then cooled to −78 °C. A solution of the crude 26 and acetaldehyde (5.0 M in THF, 1.6 mL, 8.00 mmol) in dry THF (0.5 mL) was added dropwise and the resultant solution was stirred at the same temperature for 1 h. The reaction mixture was quenched by adding H2O with stirring at r.t. for 30 min, filtered through Celite, and extracted three times with CH2Cl2. The combined organic phases were dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel to give 31 (91.7 mg, 55%, 2 steps, 31-major/31-minor ratio = 85 : 15 by 600 MHz 1H-NMR) as a colorless oil. 1H-NMR (600 MHz, CDCl3) δ: 4.14 (dq, J = 6.0, 6.0 Hz, 0.85H), 3.91 (dq, J = 6.6, 7.8 Hz, 0.15H), 3.33 (ddd, J = 6.0, 7.2, 10.8 Hz, 0.85H), 3.24 (ddd, J = 7.8, 7.8, 10.8 Hz, 0.15H), 2.01 (dd, J = 10.8, 10.8 Hz, 0.15H), 1.97 (dd, J = 10.8, 10.8 Hz, 0.85H), 1.85 (dd, J = 7.2, 10.8 Hz, 0.85H), 1.69–1.79 (m, 2.15H), 1.48–1.62 (m, 2H), 1.34–1.45 (m, 1.85H), 1.18–1.31 (m, 5.15H), 0.90 (s, 6H). 13C-NMR (150 MHz, CDCl3) δ: 218.4, 217.3, 68.3, 65.6, 62.9, 62.7, 61.5, 60.8, 35.3, 35.3, 35.1, 35.1, 29.8,* 29.5, 29.5, 28.8, 27.7, 27.5, 26.7, 26.5,* 26.1, 21.2, 21.0. HRMS-FAB (m/z): [M + H]+ Calcd for C13H23O2, 211.1698; Found, 211.1703.
*These signals might be derived from the minor conformation of cyclohexane.
2-[1-(Methoxymethoxy)ethyl]-7,7-dimethylspiro[3.5]nonan-1-one (24)To a solution of 31 (211.2 mg, 1.01 mmol) in CH2Cl2 (4.5 mL) were added N,N-diisopropylethylamine (2.1 mL, 12.0 mmol) and chloromethyl methyl ether (0.46 mL, 6.08 mmol) at 0 °C and then the solution was warmed to r.t. After stirring for 17 h, the reaction mixture was washed two times with saturated aqueous NaHCO3, dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 24 (238.3 mg, 93%, 24-major/24-minor ratio = 75 : 25 by 600 MHz 1H-NMR) as a colorless oil. 1H-NMR (600 MHz, CDCl3) δ: 4.70 (d, J = 6.6 Hz, 0.25H), 4.65 (d, J = 6.6 Hz, 0.25H), 4.65 (d, J = 6.6 Hz, 0.75H), 4.59 (d, J = 6.6 Hz, 0.75H), 4.01 (dq, J = 6.0, 6.0 Hz, 0.75H), 3.87 (dq, J = 6.0, 6.0 Hz, 0.25H), 3.42 (ddd, J = 6.0, 7.8, 10.8 Hz, 0.25H), 3.38 (s, 0.75H), 3.34 (s, 2.25H), 3.29 (ddd, J = 6.0, 7.8, 10.8 Hz, 0.75H), 1.98 (dd, J = 10.8, 10.8 Hz, 0.75H), 1.95 (dd, J = 10.8, 10.8 Hz, 0.25H), 1.90 (dd, J = 7.8, 10.8 Hz, 0.75H), 1.68–1.77 (m, 2.25H), 1.50–1.58 (m, 2H), 1.34–1.45 (m, 2H), 1.17–1.30 (m, 5H), 0.90 (s, 4.5H), 0.90 (s, 1.5H). 13C-NMR (150 MHz, CDCl3) δ: 216.8, 216.4, 95.0, 94.9, 71.5, 70.7, 62.3, 62.3, 60.2, 59.4, 55.5, 55.5, 35.3, 35.3, 35.1, 35.0, 30.0,* 29.5, 28.9, 28.8, 27.1, 26.8, 26.7, 26.7, 26.3,* 18.6, 18.3. HRMS-FAB (m/z): [M + H]+ Calcd for C15H27O3, 255.1960; Found, 255.1967. *These signals might be derived from the minor conformation of cyclohexane.
(1R*,2S*)-2-[1-(Methoxymethoxy)ethyl]-7,7-dimethylspiro[3.5]nonane-1-carbonitrile (32)To a stirred solution of 24 (87.0 mg, 0.343 mmol) in 1,2-dimethoxyethane (34.0 mL) were added p-toluenesulfonylmethyl isocyanide (268.0 mg, 1.37 mmol) and t-BuOH (3.4 mL) at r.t. and the mixture was cooled to −30 °C. To the mixture was added t-BuOK (115.7 mg, 1.03 mmol) and the resultant solution was stirred at the same temperature for 30 min. Additional amounts of t-BuOK (154.3 mg, 1.38 mmol) were then added to the solution and the mixture was allowed to warm to r.t. and stirred for 30 min before heating at 50 °C. After stirring at 50 °C for 16 h, the reaction mixture was cooled to r.t., quenched with saturated aqueous NH4Cl and extracted three times with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 32 (32.0 mg, 35%, 32-major/32-minor ratio = 60 : 40 by 600 MHz 1H-NMR) as a colorless oil. 1H-NMR (600 MHz, CDCl3). δ 4.73 (d, J = 7.2 Hz, 0.4H), 4.70 (d, J = 7.2 Hz, 0.6H), 4.60 (d, J = 7.2 Hz, 0.6H), 4.59 (d, J = 7.2 Hz, 0.4H), 3.67 (dq, J = 6.0, 6.0 Hz, 0.6H), 3.59 (dq, J = 6.0, 6.0 Hz, 0.4H), 3.40 (s, 1.2H), 3.38 (s, 1.8H), 2.66 (d, J = 9.0 Hz, 0.4H), 2.63 (d, J = 9.0 Hz, 0.6H), 2.47–2.55 (m, 1H), 1.91 (dd, J = 9.0, 10.8 Hz, 0.6H), 1.88 (dd, J = 10.2, 10.8 Hz, 0.4H), 1.77–1.85 (m, 1H), 1.56–1.70 (m, 2.6H), 1.49–1.54 (m, 0.4H), 1.35–1.47 (m, 2H), 1.15–1.31 (m, 3H), 1.12 (d, J = 6.0 Hz, 1.2H), 1.12 (d, J = 6.0 Hz, 1.8H), 0.91 (s, 3H), 0.89 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 120.5,* 120.1, 95.1, 95.1, 74.8, 73.6, 55.7, 55.5, 40.4, 40.2, 38.4, 38.1, 38.0,* 37.6,* 37.1, 36.3, 35.5, 35.5, 35.4, 35.3, 35.2, 34.9, 33.6, 33.4, 33.3,* 32.9,* 32.4,* 32.4,* 32.1,* 31.7,* 31.0,* 30.0, 29.9, 29.5, 29.4, 29.4, 25.2,* 18.4,* 17.5,* 17.5, 17.3. HRMS-FAB (m/z): [M + H]+ Calcd for C16H28NO2, 266.2120; Found, 266.2137. *These signals might be derived from the minor conformation of cyclohexane.
1-{(1R*,2S*)-2-[1-(Methoxymethoxy)ethyl]-7,7-dimethylspiro[3.5]nonan-1-yl}-2-phenylethan-1-oneTo a stirred solution of benzyl magnesium chloride (1.0 M in Et2O, 0.64 mL, 0.640 mmol) in Et2O (0.5 mL) was added 32 (42.3 mg, 0.160 mmol) in Et2O (2.0 mL) at 0 °C and the solution was refluxed for 3.5 h. Then 1N HCl (1.0 mL) was added and the reaction mixture was stirred at 50 °C for an additional 1 h. The resultant solution was cooled to r.t. and extracted three times with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 1-{(1R*,2S*)-2-[1-(methoxymethoxy)ethyl]-7,7-dimethylspiro[3.5]nonan-1-yl}-2-phenylethan-1-one (54.4 mg, 95%, major/minor ratio = 60 : 40 by 600 MHz 1H-NMR) as a colorless oil. 1H-NMR (600 MHz, CDCl3) δ: 7.29–7.34 (m, 2H), 7.18–7.27 (m, 3H), 4.58 (d, J = 6.6 Hz, 0.6H), 4.58 (d, J = 6.6 Hz, 0.4H), 4.45 (d, J = 6.6 Hz, 0.6H), 4.44 (d, J = 6.6 Hz, 0.4H), 3.57–3.70 (m, 2H), 3.46 (dq, J = 6.0, 6.0 Hz, 0.4H), 3.41 (dq, J = 6.6, 6.6 Hz, 0.6H), 3.25 (s, 1.8H), 3.23 (s, 1.2H), 2.96 (d, J = 9.0 Hz, 0.4H), 2.89 (d, J = 9.6 Hz, 0.6H), 2.65–2.73 (m, 1H), 1.83 (dd, J = 9.6, 10.8 Hz, 0.6H), 1.77 (dd, J = 10.2, 10.2 Hz, 0.4H), 1.66–1.72 (m, 0.6H), 1.58–1.64 (m, 0.4H), 1.43–1.51 (m, 2.6H), 1.36 (dd, J = 10.2, 10.8 Hz, 0.4H), 1.11–1.27 (m, 5H), 0.96 (d, J = 6.0 Hz, 1.2H), 0.90 (d, J = 6.6 Hz, 1.8H), 0.86 (s, 1.8H), 0.85 (s, 3H), 0.81 (s, 1.2H). 13C-NMR (150 MHz, CDCl3) δ: 206.7, 206.5, 133.9, 133.8, 129.7, 129.6, 128.7, 128.6, 127.0, 126.9, 95.0, 94.9, 75.9, 75.1, 56.0, 55.5, 55.2, 55.2, 51.3, 51.2, 41.1, 40.8, 36.3, 36.2, 35.5, 35.3, 34.8, 32.2,* 31.1, 30.9, 29.7, 29.7, 28.0, 27.9, 24.2,* 17.6, 17.4. HRMS-FAB (m/z): [M + H]+ Calcd for C23H35O3, 359.2586; Found, 359.2588. *These signals might be derived from the minor conformation of cyclohexane.
1-{(1R*,2S*)-2-[1-(Methoxymethoxy)ethyl]-7,7-dimethylspiro[3.5]nonan-1-yl}-2-phenylethan-1-ol (33)To a stirred solution of the above ketone (56.8 mg, 0.159 mmol) in MeOH (3.0 mL) was added NaBH4 (24.6 mg, 0.647 mmol) at 0 °C and the solution was warmed to r.t. NaBH4 (27.1 mg, 0.713 mmol and 22.4 mg, 0.589 mmol) was added to the mixture after 4 and 15 h, then the solution was stirred for an additional 6 h. The resultant mixture was quenched with saturated aqueous NH4Cl and extracted three times with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 33 (56.5 mg, 99%, 33-major/33-minor ratio = 65 : 35 by 600 MHz 1H-NMR) as a colorless solid. 1H-NMR (600 MHz, CDCl3) δ: 7.29–7.33 (m, 2H), 7.20–7.24 (m, 3H), 4.75 (d, J = 6.6 Hz, 0.65H), 4.70 (d, J = 6.6 Hz, 0.35H), 4.66 (d, J = 6.6 Hz, 0.65H), 4.63 (d, J = 6.6 Hz, 0.35H), 3.83–3.89 (m, 1H), 3.72 (dq, J = 3.0, 6.0 Hz, 0.65H), 3.64 (m, 0.35H), 3.43 (s, 1.95H), 3.37 (s, 1.05H), 3.12 (dd, J = 2.4, 13.8 Hz, 0.35H), 2.87 (dd, J = 2.4, 13.8 Hz, 0.65H), 2.40 (dd, J = 10.8, 13.8 Hz, 0.65H), 2.37 (dd, J = 10.8, 13.8 Hz, 0.35H), 2.12–2.23 (m, 0.35H), 2.02–2.09 (m, 1H), 1.82–1.87 (m, 0.65H), 1.63–1.80 (m, 3H), 1.49–1.57 (m, 1.65H), 1.37–1.44 (m, 1.35H), 1.21–1.34 (m, 3H), 1.10–1.20 (m, 5H), 0.86 (s, 3H), 0.84 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 139.3, 138.8, 129.4, 129.4, 128.6, 128.6, 126.5, 126.3, 95.4, 95.2, 77.8, 74.8,* 73.6, 73.4, 55.6, 55.5, 54.1, 50.1, 42.6, 42.4, 38.3, 37.8, 37.7, 37.4, 37.3, 36.6, 35.7, 35.6, 35.3, 35.2, 35.1, 34.9, 33.0,* 31.6, 30.0, 30.0, 29.7,* 28.9, 27.6, 27.4, 23.8,* 18.0, 17.5,* 17.1. HRMS-FAB (m/z): [M + H]+ Calcd for C23H37O3, 361.2743; Found, 361.2744. *These signals might be derived from the minor conformation of cyclohexane.
(1R*,2S*)-2-[1-(Methoxymethoxy)ethyl]-7,7-dimethyl-1-[(E)-styryl]spiro[3.5]nonane (34)To a stirred solution of 33 (53.8 mg, 0.149 mmol) in CH2Cl2 (2.5 mL) were added Et3N (0.13 mL, 0.940 mmol) and methanesulfonyl chloride (0.07 mL, 0.901 mmol) at 0 °C and the solution was stirred at the same temperature for 14 h. The resultant mixture was washed two times with saturated aqueous NaHCO3 and once with brine, dried over Na2SO4 and concentrated. This crude mesylate was used in the next step without further purification. To a solution of the crude mesylate in DMSO (3.0 mL) was added t-BuOK (67.8 mg, 0.605 mmol) at r.t. After stirring for 10 min, the resultant mixture was quenched with saturated aqueous NH4Cl and extracted five times with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 34 (36.9 mg, 72%, 2 steps, 34-major/34-minor ratio = 65 : 35 by 600 MHz 1H-NMR) as a colorless solid. 1H-NMR (600 MHz, CDCl3) δ: 7.34–7.38 (m, 2H), 7.26–7.31 (m, 2H), 7.16–7.21 (m, 1H), 6.39 (d, J = 15.6 Hz, 0.35H), 6.34 (d, J = 15.6 Hz, 0.65H), 6.31 (dd, J = 8.4, 15.6 Hz, 0.35H), 6.27 (dd, J = 8.4, 15.6 Hz, 0.65H), 4.71 (d, J = 6.6 Hz, 0.65H), 4.65 (d, J = 6.6 Hz, 0.35H), 4.61 (d, J = 6.6 Hz, 0.65H), 4.59 (d, J = 6.6 Hz, 0.35H), 3.59–3.66 (m, 1H), 3.39 (s, 1.95H), 3.34 (s, 1.05H), 2.53 (dd, J = 8.4, 8.4 Hz, 0.35H), 2.40 (dd, J = 8.4, 8.4 Hz, 0.65H), 2.26–2.33 (m, 0.35H), 2.21 (m, 0.65H), 1.94 (dd, J = 8.4, 10.8 Hz, 0.65H), 1.81 (dd, J = 9.0, 10.8 Hz, 0.35H), 1.48–1.62 (m, 3H), 1.44 (dd, J = 9.6, 10.8 Hz, 0.65H), 1.16–1.37 (m, 5.35H), 1.09 (d, J = 6.6 Hz, 1.05H,), 1.05 (d, J = 6.6 Hz, 1.95H), 0.86 (s, 3H), 0.81 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 138.0, 137.8, 131.3, 130.7, 130.3, 129.9, 128.5, 128.4, 126.9, 126.7, 126.1, 95.0, 94.9, 76.8,* 55.4, 55.3, 51.4, 50.7, 41.4, 41.2, 40.6, 40.5, 36.3, 36.2, 35.6, 35.6, 35.3, 35.3, 33.1, 32.1, 31.8,* 29.9, 28.6, 28.5, 25.0,* 18.3, 17.9. HRMS-FAB (m/z): [M + H]+ Calcd for C23H35O2, 343.2637; Found, 343.2640. *These signals might be derived from the minor conformation of cyclohexane.
1-{(1R*,2S*)-7,7-Dimethyl-1-[(E)-styryl]spiro[3.5]nonan-2-yl}ethan-1-olTo a stirred solution of 34 (53.8 mg, 0.157 mmol) in MeOH (4.5 mL) was added 6N HCl (1.5 mL, 9.00 mmol) at r.t. and the mixture was stirred for 6 h. The resulting solution was quenched with saturated aqueous NaHCO3 and extracted five times with CH2Cl2. The combined organic phases were dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 1-{(1R*,2S*)-7,7-dimethyl-1-[(E)-styryl]spiro[3.5]nonan-2-yl}ethan-1-ol (44.5 mg, 95%, major/minor ratio = 65 : 35 by 600 MHz 1H-NMR) as a colorless solid. 1H-NMR (600 MHz, CDCl3) δ: 7.34–7.38 (m, 2H), 7.27–7.32 (m, 2H), 7.18–7.22 (m, 1H), 6.44 (d, J = 15.6 Hz, 0.35H), 6.36 (d, J = 15.6 Hz, 0.65H), 6.35 (dd, J = 8.4, 15.6 Hz, 0.35H), 6.27 (dd, J = 8.4, 15.6 Hz, 0.65H), 3.69–3.78 (m, 1H), 2.51 (dd, J = 8.4, 8.4 Hz, 0.35H), 2.40 (dd, J = 8.4, 8.4 Hz, 0.65H), 2.13–2.24 (m, 1H), 1.90 (dd, J = 8.4, 10.8 Hz, 0.65H), 1.82 (dd, J = 8.4, 10.8 Hz, 0.35H), 1.49–1.64 (m, 3H), 1.16–1.43 (m, 6H), 1.11 (d, J = 6.6 Hz, 1.05H), 1.09 (d, J = 6.6 Hz, 1.95H), 0.86 (s, 3H), 0.81 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 137.7, 137.6, 131.2, 130.6, 130.4, 130.3, 128.5, 128.5, 127.0, 127.0, 126.1, 126.1, 73.1, 71.8, 51.4, 50.3, 42.7, 42.6, 40.4, 40.4, 36.4, 36.3, 35.6, 35.6, 35.3, 35.3, 31.9, 31.8, 31.6,* 29.9, 28.7, 28.6, 25.6,* 21.2, 20.5. HRMS-FAB (m/z): [M + Na]+ Calcd for C21H30ONa, 321.2194; Found, 321.2215. *These signals might be derived From the minor conformation of cyclohexane.
1-{(1R*,2S*)-7,7-Dimethyl-1-[(E)-styryl]spiro[3.5]nonan-2-yl}ethan-1-one (23)To a stirred solution of above secondary alcohol (9.7 mg, 0.0326 mmol) was added Dess-Martin periodinane (21.4 mg, 0.0505 mmol) at r.t. and the resulting suspension was stirred for 2 h. The reaction mixture was quenched with saturated aqueous Na2S2O3 and NaHCO3 and extracted three times with CH2Cl2. The combined organic phases were dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 23 (9.5 mg, 98%) as a colorless solid. 1H-NMR (600 MHz, CDCl3) δ: 7.37–7.40 (m, 2H), 7.30–7.34 (m, 2H), 7.21–7.25 (m, 1H), 6.43 (d, J = 16.2 Hz, 1H), 6.34 (d, J = 9.0, 16.2 Hz, 1H), 3.12 (ddd, J = 9.0, 9.0, 9.0 Hz, 1H), 2.75 (dd, J = 9.0, 9.0 Hz, 1H), 2.07 (s, 3H), 1.84–1.91, (m, 2H), 1.52–1.59 (m, 3H), 1.34–1.39 (m, 1H), 1.15–1.31 (m, 4H), 0.87 (s, 3H), 0.82 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 209.3, 137.2, 131.5, 129.1, 128.6, 127.3, 126.2, 51.6, 46.7, 40.4, 35.8, 35.4, 35.3, 31.1, 29.8, 28.7, 28.3. HRMS-FAB (m/z): [M + H]+ Calcd for C21H29O, 297.2218; Found, 297.2219.
Model Compound 13To a flame-dried round bottom flask was added iPr2NH (0.03 mL, 0.214 mmol) and dry THF (0.8 mL). The solution was cooled to 0 °C and n-BuLi (1.6 M in hexane) was added dropwise. After stirring at the same temperature for 20 min, the reaction mixture was cooled to −78 °C. A solution of 23 (19.6 mg, 0.0662 mmol) in dry THF (1.0 mL) was then added dropwise and the mixture was brought to 0 °C with stirring for 30 min before TMSCl (0.03 mL, 0.239 mmol) was added. The reaction mixture was stirred at 0 °C for 45 min, then quenched with saturated aqueous NH4Cl at 0 °C and extracted three times with a 1 : 1 solution of hexane : Et2O. The combined organic phases were washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4 and concentrated. This material (silyl enol ether 35) was used in the next step without further purification. To a solution of the above residue in dry CH2Cl2 (1.0 mL), a solution of aldehyde 22 (22.2 mg, 0.159 mmol) in CH2Cl2 (1.0 mL) was added dropwise at −50 °C. BF3·OEt2 (0.02 mL, 0.160 mmol) was then added dropwise and the solution was stirred at the same temperature for 3 h. The reaction mixture was quenched by adding saturated aqueous NaHCO3 at −50 °C and extracted three times with CH2Cl2. The combined organic phases were dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel to give 13 (5.5 mg, 19%) as a mixture of four diastereomers, which were purified by chiral HPLC (CHIRAL ART Cellulose-SC, YMC) to afford syn-13a, syn-13b, anti-13a, and anti-13b (isolation ratio = 16 : 16 : 34 : 34) as colorless solids, respectively.
C-6,8-syn-13a (or 13b): 1H-NMR (600 MHz, CDCl3) δ: 7.36–7.39 (m, 2H), 7.30–7.34 (m, 2H), 7.21–7.25 (m, 1H), 6.86 (ddd, J = 3.6, 4.2, 9.6 Hz, 1H), 6.42 (d, J = 15.6 Hz, 1H), 6.31 (dd, J = 9.0, 15.6 Hz, 1H), 5.99 (ddd, J = 1.8, 1.8, 9.6 Hz, 1H), 4.63–4.69 (m, 1H), 4.25–4.31 (m, 1H), 3.37 (d, J = 3.0 Hz, 1H), 3.15 (ddd, J = 9.0, 9.0, 9.0 Hz, 1H), 2.77 (dd, J = 9.0, 9.0 Hz, 1H), 2.61 (dd, J = 3.6, 18.0 Hz, 1H), 2.55 (dd, J = 9.0, 18.0 Hz, 1H), 2.39–2.43 (m, 2H), 1.96–2.02 (m, 1H), 1.85–1.93 (m, 2H), 1.76 (ddd, J = 4.2, 6.0, 13.8 Hz, 1H), 1.52–1.60 (m, 3H), 1.35–1.40 (m, 1H), 1.16–1.32 (m, 4H), 0.87 (s, 3H), 0.82 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 212.1, 164.1, 145.1, 131.8, 128.6, 127.5, 126.3, 121.3, 75.4, 64.3, 51.5, 47.1, 46.5, 40.6, 40.6, 35.4, 29.1. HRMS-FAB (m/z): [M + H]+ Calcd for C28H37O4, 437.2692; Found, 437.2691.
C-6,8-syn-13b (or 13a): 1H-NMR (600 MHz, CDCl3) δ: 7.36–7.39 (m, 2H), 7.30–7.34 (m, 2H), 7.21–7.25 (m, 1H), 6.86 (ddd, J = 3.6, 4.2, 9.6 Hz, 1H), 6.43 (d, J = 15.6 Hz, 1H), 6.30 (dd, J = 9.0, 15.6 Hz, 1H), 5.99 (ddd, J = 1.8, 1.8, 9.6 Hz, 1H), 4.63–4.69 (m, 1H), 4.25–4.31 (m, 1H), 3.31 (d, J = 3.0 Hz, 1H), 3.14 (ddd, J = 9.0, 9.0, 9.0 Hz, 1H), 2.78 (dd, J = 9.0, 9.0 Hz, 1H), 2.54–2.62 (m, 2H), 2.39–2.43 (m, 2H), 2.00 (ddd, J = 6.0, 8.4, 14.4 Hz, 1H), 1.91 (dd, J = 9.0, 10.8 Hz, 1H), 1.86 (dd, J = 9.0, 10.8 Hz, 1H), 1.75 (ddd, J = 4.2, 6.0, 14.4 Hz, 1H), 1.52–1.60 (m, 3H), 1.35–1.40 (m, 1H), 1.16–1.32 (m, 4H), 0.87 (s, 3H), 0.82 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 212.0 (no signal on 13C-NMR, however, it was observed by HMBC correlation from H-9), 164.1, 145.1, 131.9, 128.6, 127.5, 126.3, 121.3, 75.5, 64.3, 51.4, 47.0, 46.5, 40.6, 40.6, 35.4, 35.2, 29.8, 29.0. HRMS-FAB (m/z): [M + H]+ Calcd for C28H37O4, 437.2692; Found, 437.2693.
C-6,8-anti-13a (or 13b): 1H-NMR (600 MHz, CDCl3) δ: 7.39 (d, J = 1.2, 7.8 Hz, 2H), 7.30–7.34 (m, 2H), 7.23 (dt, J = 1.2, 7.8 Hz, 1H), 6.85 (ddd, J = 3.0, 6.0, 10.2 Hz, 1H), 6.43 (d, J = 15.6 Hz, 1H), 6.31 (dd, J = 9.0, 15.6 Hz, 1H), 6.00 (ddd, J = 1.2, 2.4, 9.6 Hz, 1H), 4.69–4.75 (m, 1H), 4.35–4.42 (m, 1H), 3.39 (dd, J = 1.2, 3.6 Hz, 1H), 3.12 (ddd, J = 9.0, 9.0, 9.0 Hz, 1H), 2.75 (dd, J = 9.0, 9.0 Hz, 1H), 2.58 (dd, J = 2.4, 18.0 Hz, 1H), 2.43 (dd, J = 9.6, 18.0 Hz, 1H), 2.36 (dddd, J = 1.2, 4.2, 5.4, 18.0 Hz, 1H), 2.30 (dddd, J = 2.4, 2.4, 11.4, 18.0 Hz, 1H), 1.85–1.92 (m, 2H), 1.77 (dddd, J = 1.8, 2.4, 9.0, 14.4 Hz, 1H), 1.70 (ddd, J = 3.6, 10.2, 14.4 Hz, 1H), 1.52–1.60 (m, 3H), 1.35–1.40 (m, 1H), 1.16–1.32 (m, 4H), 0.87 (s, 3H), 0.82 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 212.0, 145.1, 137.1, 131.9, 128.6, 127.5, 126.3, 121.4, 74.7, 63.7, 51.7, 47.3, 46.5, 41.6, 40.6, 35.8, 35.4, 35.2, 29.9, 29.8, 28.6, 23.0. HRMS-FAB (m/z): [M + H]+ Calcd for C28H37O4, 437.2692; Found, 437.2688.
C-6,8-anti-13b (or 13a): 1H-NMR (600 MHz, CDCl3) δ: 7.37–7.40 (m, 2H), 7.30–7.34 (m, 2H), 7.23 (dt, J = 1.2, 7.8 Hz, 1H), 6.86 (ddd, J = 3.0, 6.0, 10.2 Hz, 1H), 6.43 (d, J = 15.6 Hz, 1H), 6.31 (dd, J = 9.0, 15.6 Hz, 1H), 6.01 (ddd, J = 1.2, 2.4, 9.6 Hz, 1H), 4.70–4.75 (m, 1H), 4.35–4.41 (m, 1H), 3.37 (dd, J = 1.8, 3.6 Hz, 1H), 3.13 (ddd, J = 9.0, 9.0, 9.0 Hz, 1H), 2.76 (dd, J = 9.0, 9.0 Hz, 1H), 2.56 (dd, J = 2.4, 18.0 Hz, 1H), 2.46 (dd, J = 9.6, 18.0 Hz, 1H), 2.37–2.43 (m, 1H), 2.30 (dddd, J = 2.4, 2.4, 11.4, 18.0 Hz, 1H), 1.85–1.93 (m, 2H), 1.77 (dddd, J = 1.8, 2.4, 9.0, 14.4 Hz, 1H), 1.71 (ddd, J = 3.6, 10.2, 14.4 Hz, 1H), 1.52–1.60 (m, 3H), 1.34–1.40 (m, 1H), 1.16–1.31 (m, 4H), 0.87 (s, 3H), 0.82 (s, 3H). 13C-NMR (150 MHz, CDCl3) δ: 212.0, 164.2, 145.1, 137.1, 131.9, 128.6, 127.4, 126.3, 121.4, 74.7, 63.7, 51.5, 47.3, 46.4, 41.6, 40.6, 35.8, 35.3, 35.2, 29.9, 29.8, 28.6. HRMS-FAB (m/z): [M + H]+ Calcd for C28H37O4, 437.2692; Found, 437.2659.
We deeply appreciate the critical comments, suggestions, and editing of the manuscript by Dr. Susan L. Morris-Natschke (UNC-CH). This study was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT KAKENHI, Japan) awarded to K.N.G. (Grant Number 25293024).
The authors declare no conflict of interest.
This article contains supplementary materials.