Regular Articles
Fragment Molecular Orbital Based Affinity Prediction toward Pyruvate Dehydrogenase Kinases: Insights into the Charge Transfer in Hydrogen Bond Networks
2023 年 71 巻 4 号 p. 299-306

詳細
2023 年 71 巻 4 号 p. 299-306
The fragment molecular orbital (FMO) method is a fast quantum-mechanics method that divides systems into pieces of fragments and performs ab initio calculations. The method has been expected to improve the accuracy of describing protein-ligand interactions by incorporating electronic effects. In this article, FMO calculation with solvation methods were applied to the affinity prediction at the ATP-binding site of PDHK4. As the ionized aspartic acid lies at the center and is involved in the complex hydrogen bond networks, this system has turned out to be a difficult target to describe by traditional molecular-mechanics method. In the FMO calculation with the polarizable continuum model (PCM) solvation method, a considerable amount of charge (−0.27e) was transferred from the ionized aspartate to the surrounding residues. We found that using FMO with the PCM solvation method was important to increase the correlation, and by incorporating the ligand deformation energy, the correlation was improved to R = 0.81 for whole twelve compounds and R = 0.91 without one outlier compound.
Predicting binding affinities as well as ranking ligands to their target proteins is an important topic in computational chemistry.1,2) The computational methods that have been developed include the linear interaction energy theory,3) the combined molecular mechanics (MM) and the generalized Born/surface area (GBSA),4) and the free energy perturbation method.5–9) These methods utilize the MM force field that assigns atom types and atomic partial charges to each atom based on the bonded environment. As the assigned partial charges do not change depending on conformation and environment, these MM methods typically lack descriptions of polarization, charge transfer and many-body effects. As a result, these methods are difficult to apply to the systems with highly charged component like metals and charged residues, or to the systems with the complex electrostatic interactions like hydrogen bond networks.10–15) Although the quantum-mechanics (QM) methods have been expected to improve the accuracy of describing protein–ligand interactions by incorporating electronic effects, applying QM methods toward large biomolecular systems has been computationally quite expensive.16)
The fragment molecular orbital (FMO) method has gained attention as an accurate and rapid scoring function to calculate the binding affinity by quantum mechanics.17–31) The systems are divided into smaller pieces called fragments, and ab initio calculations are performed for each fragment and their pairs. As the method is well suitable for parallel computing, one may expect to obtain the QM calculation result in much shorter time. Although some cases have been described reasonably well by applying FMO calculations in the gas-phase,32–35) solvation effects should be considered to evaluate binding affinities, especially for those involving interactions with the ionized fragments. For QM calculations, the solvation energies were generally calculated in combination with the implicit solvation method of the polarizable continuum model (PCM)36) or the conductor-like screening model (COSMO),37–39) or by separately calculation of the solvation energy term by MM method using generalized Born/surface area (GBSA) or the Poisson–Boltzmann/surface area (PBSA) method.4) These solvation methods were expected to increase the accuracy of the FMO binding affinity calculations, and some precedent papers have reported their usefulness. For example, Watanabe et al. studied the activity cliffs of Pim1 kinase utilizing the FMO method with the PBSA (FMO + MM-PBSA),28) and Poongavanam et al. studied the metal containing active site of human immunodeficiency virus-1 RNaseH using the FMO/PCM method.29) Although these cases were described well by incorporating the solvation energy into the FMO binding affinity calculation, little is known about the differences of the effects of these solvation methods.
In this article, we compare the result of binding affinity calculations of MM and FMO with solvation methods toward the ATP-binding site of pyruvate dehydrogenase kinase (PDHK). PDHK is a member of the GHKL ATPase/kinase superfamily, and participates in regulating the oxidative decarboxylation of pyruvate to form acetyl-CoA.40–42) The sidechain of an aspartic acid residue, which is considered to exist in the carboxylate form under physiological conditions, is located at the center of the PDHK ATP binding site, and is involved in the hydrogen bond networks in the AMP-PNP bound X-ray structure43) (Fig. 1a). The adenine binding site includes four conserved water molecules, and WAT1 water molecule bridges Asp293 and the adenine ring. The WAT1 water is a structural water as it forms four hydrogen bonds with the receptor and the ligand. The other three conserved water molecules are involved in hydrogen bond networks in a deeply buried pocket. We have recently identified novel inhibitors of PDHKs at the ATP binding site by displacing these water molecules around the aspartic acid from a fragment hit44,45) (Fig. 1b). Since the ionized aspartic acid lies at the center and is involved in the complex hydrogen bond networks, this target is considered a good model system involving several electronic effects. The accuracy of binding affinity calculations was investigated by comparing the combined result of MM, FMO and implicit solvation models.
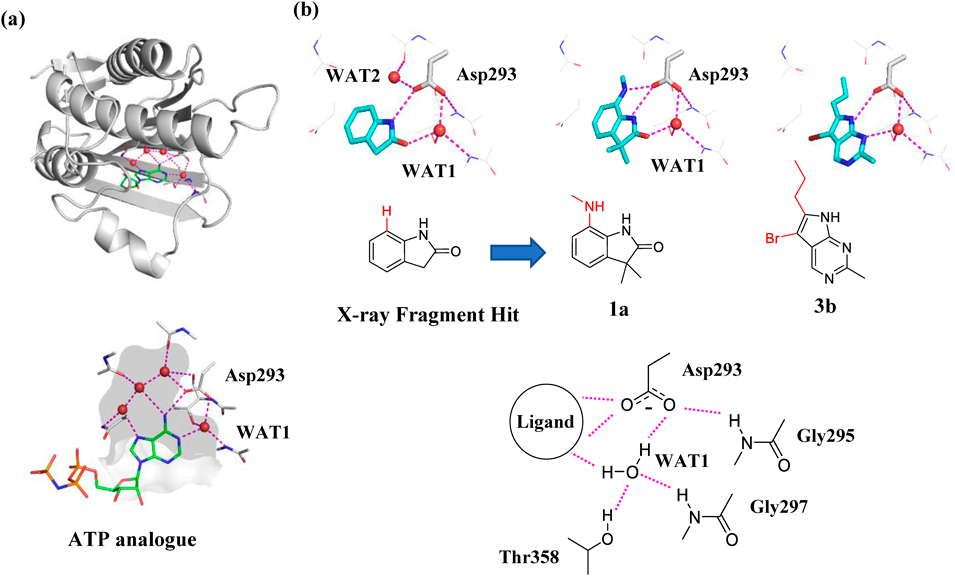
PDHKs are shown in gray cartoons, and the residues at the ATP binding site was shown in sticks in gray carbons. Water molecules are represented as red spheres, and the purple dashed lines indicate polar interactions. (a) X-ray structure of AMP-PNP with PDHK4 (PDB ID: 3CRL). AMP-PNP is shown in sticks with green carbons. Protein interiors are shown in gray surface. (b) X-ray structure of the fragment hit, compound 1a and compound 3b with PDHKs (PDB ID: 7EAT, 7EBG and 7VBU). The ligands are shown in sticks with cyan carbons. Hydrogen bond networks were shown in schematic representation.
The X-ray crystal structural data of the fragment hit with PDHK4 (Protein Data Bank (PDB) ID = 7EAT) was used as the initial geometries, and the structure was processed using Schrödinger’s protein preparation wizard (version 2021-2). All crystal waters were removed except two waters around Asp 293 (WAT1 and WAT2 in Fig. 1b), and hydrogen atoms were added using Schrödinger’s H-bond assignment/sample water orientation component. Attached hydrogen positions were energy optimized using the default protein preparation wizard settings with OPLS4 force field. For the dimethyl indole series of the reference compound and 1a–5a (Fig. 2a), the ligand initial geometries were modeled based on the ligand structure of the 7EAT PDB complex by maximizing the structural overlap. For the pyrimido indole series of compounds 1b–6b (Fig. 2b), X-ray crystal structural data of compound 3b with PDHK2 was superposed on the 7EAT PDB complex of PDHK4 by maximizing the protein backbone overlap, and the aligned position of compound 3b was used as a template for aligning other compounds. As the compounds 1a–5a and 1b–6b have substituents oriented toward WAT2 to displace this water molecule, WAT2 (Fig. 1b) was removed from the complex models of these compounds.
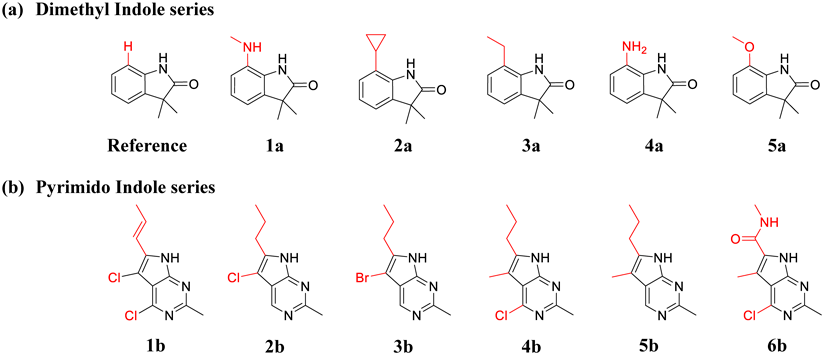
The core structures were shown in black, and the converted structures were shown in red.
The protein (including water molecules)/ligand complex models were further refined by FMO geometry optimization. Here, the protein was divided into amino acid fragments between Cα atoms and the adjacent carbonyl carbon atoms of the main chain. The protein residues which had any atom within a radius of 8 Å around the ligand (Met251–His266, Ile276–Val278, Ile289–Phe307, Gly333–Ser337, Leu348–Ile362 + water) were truncated for the geometry optimization. Both ends of the extracted amino acid fragments were capped with acylate or amide groups while maintaining the geometry of the neighboring residues. FMO geometry optimization was carried out using the General Atomic and Molecule Electrostatic Structure System (GAMESS) package (ver2018).46) The ligands and the sidechains of fragments which have any atom within a radius of 5 Å from the ligands were optimized, while other protein atoms were frozen at the experimental positions. The geometry optimization was performed at the FMO2/RHF-D method with the 6–31 G basis set until the maximum gradient became less than 3.0 × 10−4 Hartree/Bohr, using the one residue per fragment division. The FMO optimized atomic positions of the truncated structures were put back into the full system, and the merged structures were used for the next MM and FMO energy calculations.
MM and FMO Binding Energy CalculationBinding energy was obtained by performing MM/GBSA (Schrödinger ver 2021.02) and FMO-MP2/PCM calculations (GAMESS ver2018) on the truncated systems. Although the entropy plays an important role in ligand binding, as the studied twelve compounds had similar scaffolds with relatively rigid substituents, we neglected the entropy energy term on this study. Based on our recent benchmark study of FMO system truncation,47) the ligands and the amino acid fragments having any atom within a radius of 12 Å around the ligand (Val204–Leu227, Leu248–His266, Thr274–Leu282, Lys290–Ser308, Gly333–Ala341, Asp347–Tyr363 and water) were truncated for the calculations. Both ends of the extracted amino acid fragments were capped with acylate or amide groups while maintaining the geometry of the neighboring residues. The calculated binding energy of the full system and the truncated system were compared for compounds 1a and 3b (Supplementary Table S1 in Supplementary Materials). The energy differences were under 0.23 kcal/mol, suggesting that the present truncation size well reflects the calculation result of the full system.
For MM/GBSA calculations, the potential energy of each truncated complex, receptor and ligand were calculated using the Schrödinger’s Macromodel module with OPLS4 forcefield in the default settings. The binding energy of MM/GBSA (∆GbindMM/GBSA) was calculated in the supermolecular approach using the Eq. 1. For MM-PBSA calculations, the solvation energy was calculated using the PBSA_solvation_energy_calculation.svl program in the MOE package (ver 2022.02) with OPLS4 forcefield.
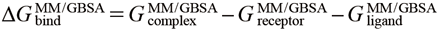 | (1) |
FMO calculations were performed at the FMO2-RHF/PCM[1(2)]/6-31G* level toward each complex, protein, and the ligand using the GAMESS package. Here, the conductor-like PCM calculation48) was applied with a water medium (ε = 78.39, solvent radius = 1.385 Å). The cavitation contributions were based on the Claverie–Pierotti method,49,50) the dispersion and repulsion were based on the Floris–Tomasi method,51) and the default GAMESS PCM settings52) were applied for other PCM conditions. For FMO fragmentation, Asp293 and water molecules were treated as a single fragment to increase the accuracy of describing hydrogen bond networks, and other residues and ligands were treated with the one residue per fragment division. The binding energy of FMO-MP2/PCM (∆GbindFMO/PCM) was calculated in the supermolecular approach following Eq. 2. The binding energy of FMO/GBSA (∆GbindFMO/GBSA) was calculated by adding the FMO interaction energy in vacuo (∆EintFMO/vacuo) and the GBSA desolvation energy (Eq. 3).
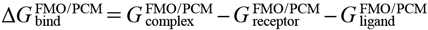 | (2) |
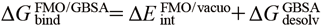 | (3) |
A two-step conformational search was carried out for twelve ligands by Schrödinger’s Macromodel module and GAMESS. In the first step, choosing OPLS4 as the force field and GBSA water model in the Macromodel, a pool of conformers was generated for each ligand in the default settings. In the second step, a pool of conformers from the above MM conformational search was energy minimized using GAMESS at the FMO2-MP2/6-31G level until the maximum gradient became less than 1.0 × 10−4 Hartree/Bohr, and the minimized conformers were energy calculated at the MP2/PCM[1(2)]/6-31G* level. The energy of the most stable conformer of each ligand was selected as the global minimum (EMP2/PCMligand min), and the FMO ligand deformation energy (∆EFMO/PCM ligand deform) was calculated from the Eq. 4. FMO binding energy with deformation energy was derived by adding the binding energy of FMO-MP2/PCM and the FMO ligand deformation energy.
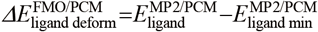 | (4) |
The analysis of the dihedral angle of amide substituents of compound 6b from the core structure were performed using the QM and MD methods. The dihedrals were scanned quantum mechanically using the GAMESS package from 0–180° with an increment of 10° at the MP2/PCM [1(2)]/6-31G* level. The dihedral populations were analyzed using the MD trajectories of the ligand free-state and the PDHK4 bound-state. MD simulations were performed with the Desmond program of Schrödinger suite with the default settings. For the dihedral analysis of the ligand free-state, the four 25 ns MD simulation were performed from both the flipped and the docking conformation of compound 6b, and the last eight 12.5 ns MD trajectories were analyzed. For the dihedral analysis of the PDHK4 bound-state, four 25 ns MD simulation were carried out with the docking model of compound 6b obtained in the previous paragraph, and the last four 12.5 ns MD trajectories were analyzed to understand the dihedral populations during the MD simulations.
Eleven PDHK4 inhibitors with experimental inhibitory activity were obtained from our previous work, and the inhibitory activity of compound 5a was also measured (Fig. 2 and Table 1). The experimentally determined IC50 activity was converted to the binding free energy using the equation ∆Gexp = RTlnKd, where R is the gas constant and T is the absolute temperature, assuming the IC50 is equivalent to Kd. The reference compound, which has no substituent oriented toward the deep binding pocket (Fig. 2a), was used as a base compound and the energy difference was derived both for real and calculated data. Although the relative experimental energy ranges up to −2.80 kcal/mol, the calculated energy in vacuo gave a much larger value, up to −30.0 kcal/mol both in the MM and FMO methods. By incorporating the solvation energy, the calculated energy approached the experimental value of up to −15.0 kcal/mol for GBSA and up to −10.0 kcal/mol for the PCM method.
| ID | Experimental affinity | Relative calculated energy | Deform energy | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (µM) | Absolute ∆Gexp | Relative ∆∆Gexp | MM/vacuo ∆∆EintMM/vac | FMO/vacuo ∆∆EintFMO/vac | MM/GBSA ∆∆GMM/GBSAbind | FMO/GBSA ∆∆GFMO/GBSAbind | FMO/PCM ∆∆GFMO/PCMbind | Ligand deformation ∆EFMO/PCMlig deform | |
| ref. | 890 | −4.17 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 3.12 |
| 1a | 12.8 | −6.69 | −2.52 | −6.26 | −11.52 | −2.84 | −8.10 | −8.25 | 5.37 |
| 2a | 43.6 | −5.96 | −1.79 | 7.13 | −4.08 | 12.11 | 0.90 | −5.62 | 5.83 |
| 3a | 304 | −4.81 | −0.64 | 10.67 | −0.87 | 12.95 | 1.41 | −2.51 | 6.43 |
| 4a | 455 | −4.57 | −0.40 | −5.08 | −10.29 | 1.84 | −3.38 | −3.40 | 3.87 |
| 5a | 842 | −4.20 | −0.03 | −3.80 | −4.22 | −4.93 | −5.35 | −4.39 | 7.82 |
| 1b | 8.00 | −6.97 | −2.80 | −16.17 | −19.26 | −5.02 | −8.11 | −8.80 | 3.94 |
| 2b | 11.0 | −6.78 | −2.61 | −10.70 | −13.09 | 0.18 | −2.21 | −7.19 | 3.56 |
| 3b | 12.4 | −6.71 | −2.54 | −11.14 | −14.21 | 0.32 | −2.75 | −8.34 | 2.88 |
| 4b | 36.7 | −6.06 | −1.89 | −11.64 | −14.85 | −1.46 | −4.67 | −6.44 | 5.53 |
| 5b | 144 | −5.25 | −1.08 | −7.93 | −10.95 | 1.93 | −1.09 | −4.91 | 3.85 |
| 6b | 198 | −5.06 | −0.89 | −30.82 | −30.96 | −14.44 | −14.58 | −10.08 | 6.11 |
Relative binding energies are from the reference compound. Experimental affinity was derived from real IC50 values. All energies are in kcal/mol. Chemical structures are shown in Fig. 2.
The calculated energies from the MM and FMO-based methods were assessed using the Pearson’s correlation coefficient (R) with respect to the experimental energy (Fig. 3).
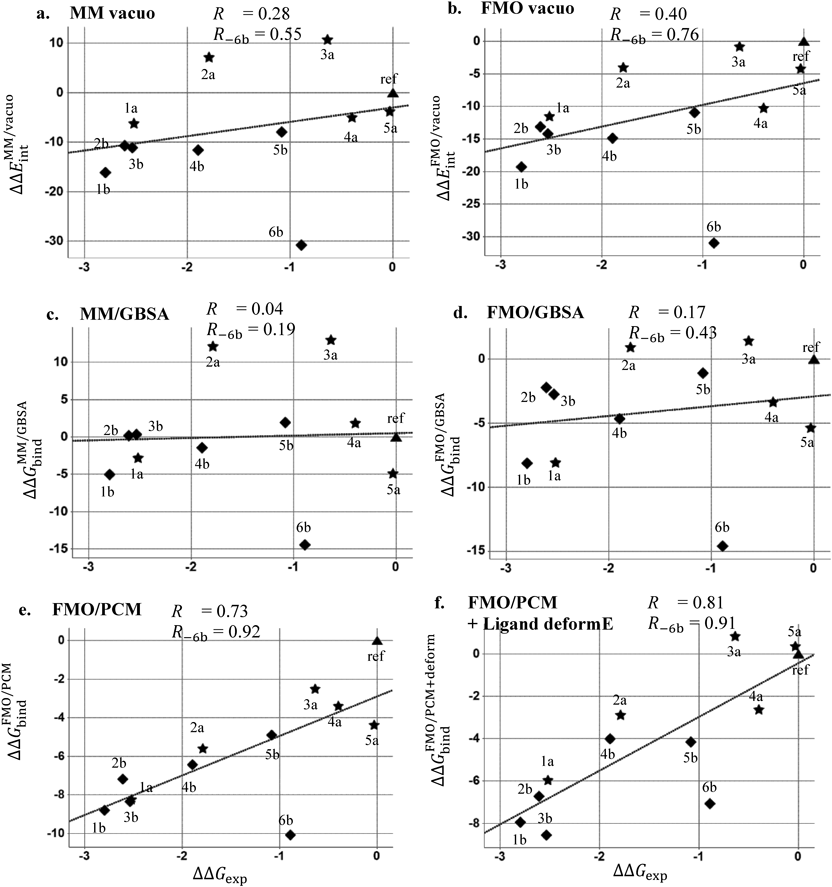
All energies are in kcal/mol. R denotes a Pearson’s correlation coefficient for all compounds, and R − 6b denotes for all compounds without 6b. The relative experimental value was plotted on horizontal axis and calculated values on vertical axis. The data of dimethyl Indole series in Fig. 2a was plotted in star shape, the pyrimido Indole series in Fig. 2b was in diamond shape and the reference compound was plotted in triangle. The regression line was shown in dot lines.
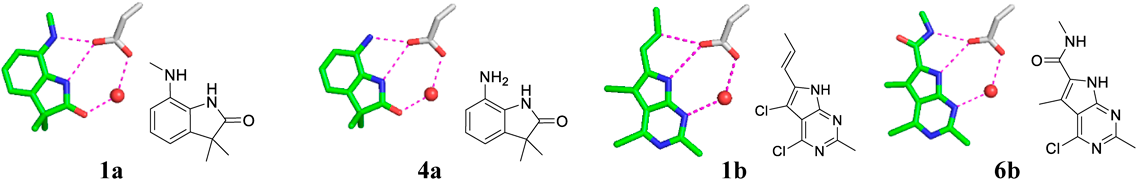
The MM interaction energy in vacuo (Fig. 3a) showed poor correlation (R = 0.28), and the FMO interaction energy in vacuo (Fig. 3b) slightly improved the correlation (R = 0.40). The interaction energy of compound 6b, which forms an additional hydrogen bond interaction with Asp293 at the center using the amide substituent in the docking model (Fig. 4), was overestimated and was far removed from the regression lines. Compound 4a also forms a similar polar interaction with Asp293 (Fig. 4), and this compound was also overestimated as compared to the compounds 2a and 3a. By removing the outlier compound 6b, the correlation was improved to 0.55 in the MM/vacuo and 0.76 in the FMO/vacuo. The effects of including solvation energy were then investigated. Although the calculated binding energy value approached the experimental value by incorporating the solvation energy term, the GBSA method worsened the correlation both in MM and FMO measurements (Figs. 3c and 3d). The effects of including the PBSA solvation energy were also investigated (Supplementary Figure S1 in Supplementary Materials). The desolvation energy calculated by the PBSA method was weaker than the GBSA method on the present PDHK4 system, and the correlation coefficient (R) of MM/PBSA and FMO + MM-PBSA was close to the in vacuo result, 0.25 and 0.42 respectively. In contrast, by adding QM-based solvation energy term with PCM in the FMO method, the correlation was improved to give R of 0.73 for all twelve compounds and R of 0.92 without the compound 6b (Fig. 3e).
The compound is shown in sticks with green carbons. Asp293 of PDHK4 at the ATP binding site is shown in sticks in gray carbons, and water molecule is represented as red spheres.
To understand the factors of differences observed on the solvation effect between the GBSA and PCM method, The relative desolvation penalty calculated by the supermolecular approach of each method was analyzed (Table 2). As was shown in Fig. 4, the compounds of 1a, 4a, 1b and 6b form additional polar interactions with Asp293 using attached substituents, and the desolvatoin penalty of the PCM method for these systems were similar or larger values to the GBSA method. In contrast, the PCM method gave smaller desolvation penalty than the GBSA method for other compounds with the hydrophobic substituents. The electrostatic interactions of compounds 1a, 4a, 1b and 6b were well penalized by the desolvation effect of the PCM method and is considered to improve the correlation.
| 1a | 2a | 3a | 4a | 5a | 1b | 2b | 3b | 4b | 5b | 6b | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GBSA | 3.42 | 4.97 | 2.28 | 6.91 | −1.13 | 11.14 | 10.88 | 11.45 | 10.18 | 9.86 | 16.37 |
| PCM | 3.25 | −1.57 | −1.66 | 6.87 | −0.18 | 10.44 | 5.88 | 5.85 | 8.38 | 6.02 | 20.85 |
All energies are in kcal/mol. Chemical structures are shown in Fig. 2.
To further understand the factors of differences observed on the solvation effect between the GBSA and PCM method, the charge transfer from a fragment of Asp293/WAT1 in the FMO/PCM calculation was then analyzed (Table 3). Although the total formal charge of Asp293/WAT1 fragment was −1e in the MM calculation, a considerable amount of charge was transferred from this fragment in the FMO/PCM calculation. Around −0.27e charge was transferred to the surrounding fragments in the complex state, and −0.22e charge was transferred at the receptor state. Most of the charge was distributed to the fragments of the ligand, Gly295, Gly297 and Thr358 in the hydrogen bond networks (Fig. 1b, schematic diagram) with the Asp293/WAT1 fragment, resulting in a charge state quite different from the MM calculation. Besides, the amount of charge transfer from Asp293/WAT1 fragment to the ligand is different depending on the interaction. The compounds 1a, 4a, 1b and 6b formed additional hydrogen bond interactions with Asp293 using the attached substituents (Fig. 4), and these ligands accepted more than −0.050e charge from the Asp293/WAT1 fragment, values which are larger than obtained with other ligands. This means that the polarization state of the complex differs depending on the binding ligands. A considerable amount of charge transfer from the Asp293/WAT1 fragment to the surrounding residues and the difference of polarization state based on the binding ligands was observed in the FMO/PCM calculation. This suggests that describing these electronic effects through QM based solvation method is necessary to describe the energy term of the PDHK4 system with the hydrogen bond networks around the charged aspartate residue.
| (a) Charge transfer in complex state | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | 1a | 2a | 3a | 4a | 5a | 1b | 2b | 3b | 4b | 5b | 6b |
| Asp293/WAT1 | +0.265 | +0.272 | +0.272 | +0.268 | +0.279 | +0.276 | +0.273 | +0.274 | +0.274 | +0.269 | +0.273 |
| Ligand | −0.056 | −0.048 | −0.049 | −0.056 | −0.047 | −0.050 | −0.047 | −0.049 | −0.043 | −0.039 | −0.061 |
| Gly295 | −0.059 | −0.060 | −0.061 | −0.057 | −0.060 | −0.058 | −0.058 | −0.058 | −0.058 | −0.059 | −0.057 |
| Gly297 | −0.040 | −0.041 | −0.041 | −0.041 | −0.041 | −0.042 | −0.040 | −0.040 | −0.043 | −0.041 | −0.040 |
| Thr358 | −0.085 | −0.088 | −0.087 | −0.089 | −0.103 | −0.096 | −0.095 | −0.095 | −0.096 | −0.096 | −0.093 |
| (b) Charge transfer in receptor state | |||||||||||
| Compound | 1a | 2a | 3a | 4a | 5a | 1b | 2b | 3b | 4b | 5b | 6b |
| Asp293/WAT1 | +0.223 | +0.224 | +0.225 | +0.220 | +0.222 | +0.225 | +0.222 | +0.222 | +0.224 | +0.222 | +0.225 |
| Gly295 | −0.063 | −0.062 | −0.063 | −0.060 | −0.061 | −0.061 | −0.060 | −0.060 | −0.059 | −0.060 | −0.062 |
| Gly297 | −0.038 | −0.039 | −0.038 | −0.038 | −0.038 | −0.039 | −0.038 | −0.038 | −0.040 | −0.038 | −0.039 |
| Thr358 | −0.085 | −0.086 | −0.085 | −0.088 | −0.087 | −0.090 | −0.090 | −0.090 | −0.090 | −0.090 | −0.089 |
Chemical structures are shown in Fig. 2.
Since compound 6b was still overestimated in the FMO/PCM energy calculation (Fig. 3e), another factor of binding was investigated. In the docking conformation of compound 6b, two NH hydrogen bond donors are on the same side to form a polar interaction with Asp293 (Fig. 4). By contrast, in the free state, the ligand is considered to exist also in the flipped conformation to form an intramolecular hydrogen bond interaction through the amide carbonyl and the NH of the core structure. To check which conformation is stable in the free state, the conformational search calculation around the amide substituent of compound 6b was carried out (Fig. 5A). The flipped conformation gave a more stable potential energy than the binding conformation and around 5.0 kcal/mol energy barrier was observed between the binding and the flipped conformation. Based on this observation, the ligand deformation energy was calculated at the QM method for each ligand and the energy was added to the FMO/PCM binding energy (Fig. 3f). By incorporating the ligand deformation energy, the compound 6b approached the regression lines and the combined energy improved the correlation of R from 0.73 to 0.81.
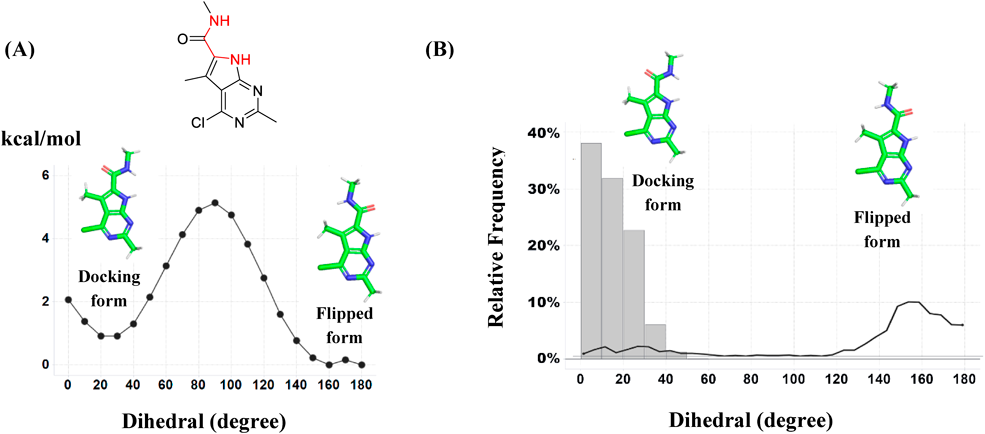
The analyzed dihedral angles are colored in red. The compound is shown in sticks with green carbons. (A) The rotational energy barrier around the amide substituent calculated by the QM method (MP2/PCM/6-31G*). (B) The dihedral population of the MD trajectories. The gray bars show the dihedral population of the complex-state MD simulation, and the black line shows the free-state MD simulation.
Figure 5B shows the dihedral population of the amide substituent of compound 6b in the MD simulation. In the MD simulation of the PDHK4 bound-state, the dihedral angle of the amide substituent was mostly between 0–30°, 14.8° on average (gray bars in Fig. 5B). In the free-state, reflecting the high energy penalty between the binding and the flipped conformation, the dihedral angle was mainly in the flipped conformation (black lines in Fig. 5B). In the present study, we neglected the entropy energy term because the structural degrees of freedom were considered almost identical for the studied twelve compounds. Based on the MD simulation result of the dihedral population of compound 6b, the binding free energy would be more reliably estimated by incorporating the entropic energy term.
FMO calculation with solvation methods were applied to the affinity prediction toward the hydrogen bond networks on the ATP-binding site of PDHK4. At this site, the sidechain of the charged aspartate locates at the center to form complex hydrogen bond networks with surrounding structural waters and was difficult to describe by the traditional MM method. We found that using FMO with the PCM solvation energy term was important to increase the correlation (R = 0.73) on this complex charged hydrogen bond networks. By incorporating ligand deformation energy as a correction term, the correlation was improved to R = 0.81 for whole twelve compounds and R = 0.91 without one outlier compound. To understand the factors of differences observed on the solvation effect between the GBSA and PCM methods, the charge transfer from a fragment of Asp293/WAT1 in the FMO/PCM calculation was analyzed. A considerable amount of charge transfer was observed toward the surrounding residues which was up to −0.27e charge in the complex state. Besides, the amount of charge transfer from the Asp293/WAT1 fragment to the ligand differed depending on the interaction pattern which was supposed to induce a different polarization state in each complex system. The obtained information suggests that describing these electronic effects through the QM based solvation method is necessary to effectively describe the energy term of the complex hydrogen bond networks with charged residues.
The authors thank Mr. Eita Nagao and Mitsumasa Takahashi for analytical support of compound 5a.
The authors declare no conflict of interest.
This article contains supplementary materials.