| To whom correspondence should be addressed: Akio Toh-e, Department of Biological Sciences, Graduate School of Science, The University of Tokyo, Hongo, Tokyo 113-0033, Japan. Tel & Fax: +81–3–5684–9420 Abbreviations: BSA, bovine serum albumin; CDK, cyclin dependent protein kinase; 3-AT, 3-amino-1,2,4-triazole; DAPI, 4',6-diamidino-2-phenylindole; DTT, dithiothreitol; EGTA, ethylene-bis-(oxyethylenenitrilo)tetraacetic acid; GFP, green fluorescent protein; LB, Luria Bertani broth; PAGE, polyacrylamide gel electrophoresis; PBS, phosphate buffered saline; PCR, polymerase chain reaction; PMSF, phenylmethylsulfonylfluoride; SC, synthetic complete medium; YPD, Yeast extract-peptone-dextrose medium. |
In Saccharomyces cerevisiae, a filamentous structure called a neck filament was observed by electron microscopy at the bud neck, which is the cleavage site at mitosis (Byers and Goetsch, 1976a). This neck filament is ca. 10 nm in diameter and is localized just under the plasma membrane. The mutation of a set of S. cerevisiae genes, CDC3, CDC10, CDC11, CDC12, and SHS1 (Haarer and Pringle, 1987; Ford and Pringle, 1991; Kim et al, 1991; Mino et al., 1998) has been known to cause a defect in cytokinesis and the disappearance of the neck filament (Byers and Goetsch, 1976b); this is consistent with the idea that these gene products are constituents of the 10 nm filament. The amino acid sequence of the gene product deduced from the nucleotide sequence of each of these genes is similar to each other and these proteins are collectively designated as septins (Haarer and Pringle, 1987; Ford and Pringle, 1991; Kim et al., 1991). Two other septins, Spr3 and Spr28, are spore-specific (Ozsarac et al., 1995; De Virgilio et al., 1996; Carroll et al., 1998). Except for Spr3 and Spr28, the septins are expressed during vegetative growth and are localized to the bud neck during the budding cycle.
Cyclins and cyclin-dependent kinase (CDK) form active kinase complexes that induce cell cycle specific events (Norbury and Nurse, 1992). In S. cerevisiae, a new bud that emerges from a mother cell grows in a polar manner after which the bud grows in an isotropic manner. This switch in growth manner is induced by the activation of mitotic CDK, primarily the Clb2/Cdc28 complex (Lew and Reed, 1993). This complex is regulated negatively by Swe1 kinase, which is a homologue of Wee1 kinase of Schizosaccharomyces pombe and is negatively regulated by Nim1 kinase (Russell and Nurse, 1987; Coleman et al., 1993). The following genes are known as genes encoding the negative regulators of Swe1 kinase: the septin genes (CDC3, CDC10, CDC11, CDC12 and SHS1), ELM1, NAP1, CLA4, HSL7 and the Nim1-related kinase genes (GIN4, HSL1 and KCC4) (Sreenivasan and Kellogg, 1999; Barral et al., 1999; Longtine et al., 2000; Shulewitz et al., 1999; Burton and Solomon, 2000). In a mutant of any of these genes, most probably due to the activation of Swe1 kinase, a bud retain its polar growth and consequently it becomes elongated (Altman and Kellogg, 1997; Tjandra et al., 1998). The pathway comprising these genes is called the mitotic signaling network. Gene products of the mitotic signaling network so far identified are localized to the bud neck in a septin-dependent manner. The question of how these proteins work for to achieve proper bud growth is one that has attracted the interest of many investigators.
Since Swe1 is a key regulator of the bud growth pattern, the functional analyses of Nim1-related kinases have been highlighted. In contrast to Nim1 of Schizosaccharomyces pombe, Nim1-related kinases of S. cerevisiae (Gin4, Hsl1 and Kcc4) are co-localized with septins at the bud neck (Tanaka and Nojima, 1996; Longtine et al., 1998; Barral et al., 1999; Okuzaki and Nojima, 2001) and have no major role in mitosis. Among Nim1-related kinases of S. cerevisiae, Gin4 has been the most extensively studied (Altman and Kellogg, 1997; Sreenivasan and Kellogg, 1999; Tjandra et al., 1998; Carroll et al., 1998; Longtine et al., 2000; Mortensen et al., 2002). Mortensen et al. (2002) revealed a spatio-temporal sequence of Gin4 interaction with its regulators that leads to its dimerization and hyperphosphorylation. Although it is well documented that Nim1-related kinase genes are needed for normal bud growth control, whether or not kinase activities are necessary for this phenomenon remains to be elucidated.
In our previous work, Nis1 was isolated by a two-hybrid screening using Shs1 as bait, and was found to be localized to the bud neck in M phase and was able to bind with all septins, Nap1, Gin4 and Kcc4 (Iwase and Toh-e, 2001). Thus, Nis1 was suggested to have some relation with the mitotic signaling network, but the exact nature of this function of Nis1 has yet to be fully elucidated.
In this study, we isolated an uncharacterized gene, YBR267w, designated as REI1 by two-hybrid screening using Nis1 as bait. We analyzed the interaction between Rei1 and other members of the mitotic signaling network. Here we report that Rei1 is cytoplasmic protein and is a member of the mitotic signaling network. Furthermore, we describe that Rei1 suppresses Gin4 kinase activity in the absence of Nap1, probably both by reducing Gin4 protein level and by inhibiting Gin4 kinase activity. We present evidence showing that Gin4 without kinase activity can support normal bud growth.
Yeast strains used in this study are listed in Table I. Yeast cells were grown on rich medium (YPD) that consisted of 2% Polypepton (Nihon Seiyaku), 1% Bacto yeast extract (Difco), 2% glucose, 0.04% adenine sulfate and 0.005% uracil or on synthetic complete medium (SC) that consisted of 0.67% yeast nitrogen base without amino acids (Difco), 2% glucose and appropriate supplements (Sherman et al., 1983). Omission medium was prepared by subtracting the indicated nutrient from SC and designated as SC-LEU, that is, the missing leucine, for example. Sporulation medium consisted of 1% potassium acetate. Medium for a two-hybrid screening was prepared by adding 30 mM 3-amino-1,2,4-triazole (3-AT) into SC-LEU-TRP-HIS medium. Standard yeast genetic manipulations were described previously (Sherman et al., 1983). Yeast transformations were performed by the lithium acetate method (Ito et al., 1983; Gietz and Schiestl, 1991). Escherichia coli DH5α (supE44 ΔlacU169 (φ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1, Hanahan, 1983) was used for construction and propagation of plasmids. E. coli BL21 (F– ompT hsdSB (rB–mB–) gal dcm, Novagen) was used for expression of T7-Rei1-6xHis. E. coli cells were grown in Luria Bertani broth (LB) that consisted of 0.8% Bacto tryptone (Difco), 0.5% Bacto-yeast extract (Difco), and 0.5% NaCl. Sodium ampicillin (40 mg/l) was supplemented to LB medium as appropriate. E. coli was grown at 37°C. E. coli transformation was carried out as described by Inoue et al. (1990). Agar was added (2%) to prepare solid media. Plasmids used in this study are listed in Table II.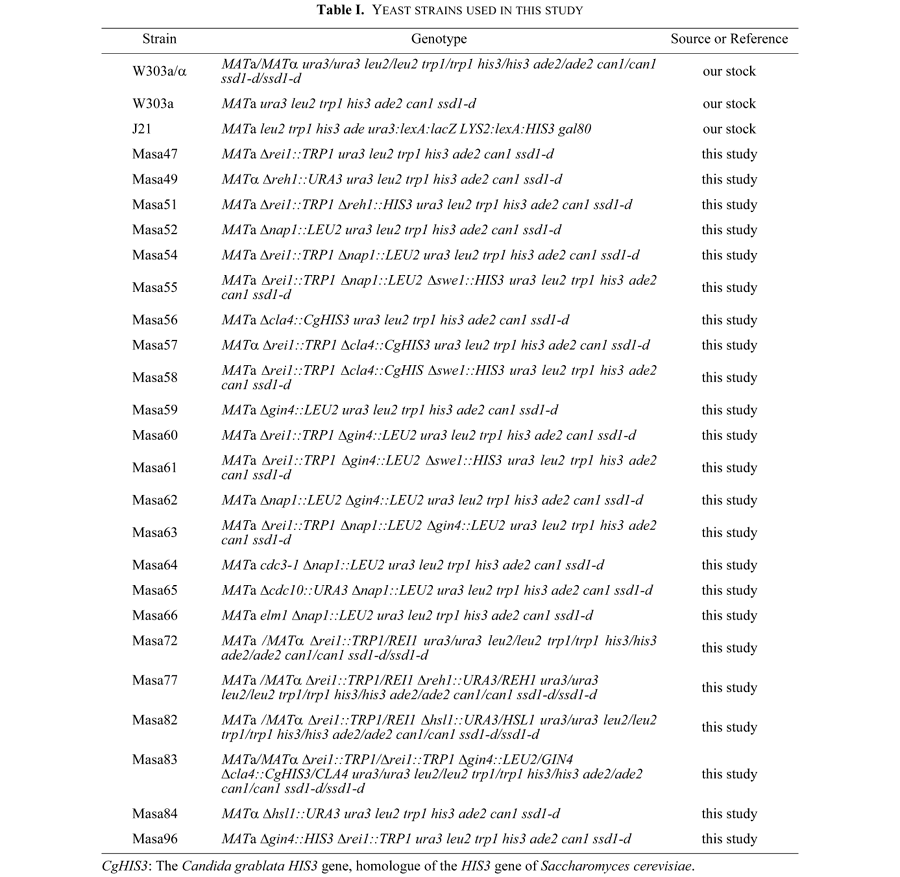
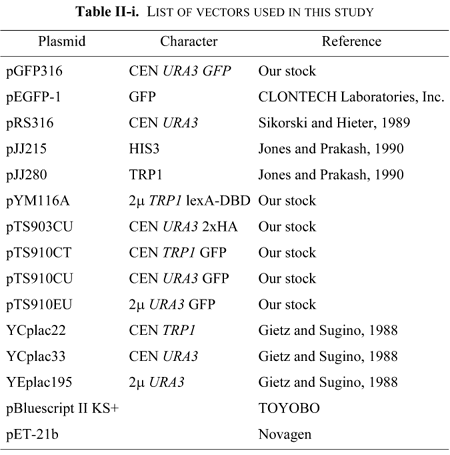
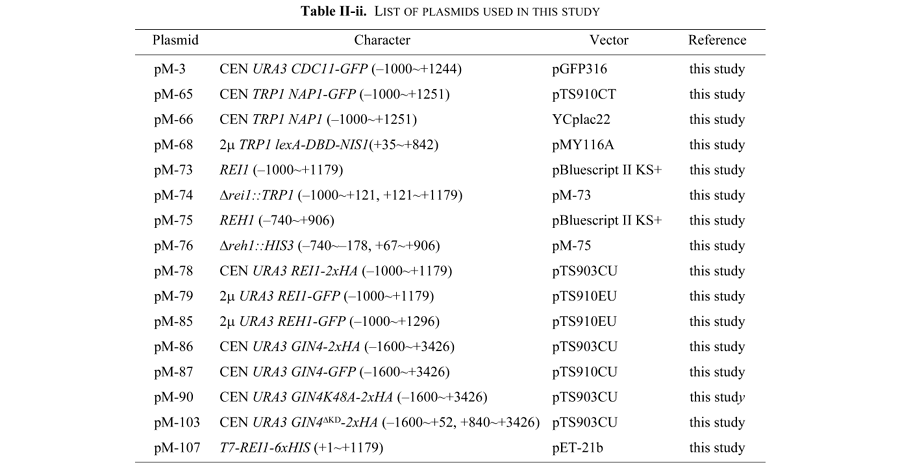
DNA preparation, restriction enzyme analysis, and agarose gel electrophoresis were carried out as described by Sambrook et al. (1989). Polymerase chain reaction (PCR) was performed using Expand high Fidelity PCR System (Roche Diagnostics Corporation) according to manufacturer's instructions. Activity of a gene cloned by the PCR method was confirmed by its ability to complement the defect of the gene of interest. Nucleotide sequences were determined by the dideoxy chain termination method (Sanger et al., 1977) using an automated DNA sequencer (ABI 370A, Applied Biosystems). The adenine nucleotide of ATG corresponding to the putative initiation codon is defined as the +1 nucleotide.
Two-hybrid analysis was performed using strain J21 containing the LexA DBD fusion plasmid and AD fusion plasmid. The extent of two-hybrid interaction was estimated by assaying β-galactosidase activity in each Leu+ Trp+ transformant. β-galactosidase activity was expressed as described (Guarente, 1983).
Cells were cultured to mid logarithmic phase and washed with phosphate buffered saline (PBS: 140 mM NaCl, 2.7 mM KCl, 3.8 mM Na2HPO4) and suspended in PBS. Fluorescence of GFP was observed with an epifluorescence microscope, IX70 (Olympus) or BX60 (Olympus) and photographed with a chilled CCD camera, SENSYS III (Nippon Roper) or PM-C35DX (Olympus) or a high resolution digital microscope, VH-8000 (Keyence).
About 108 cells were disrupted with glass beads in Lysis buffer (50 mM Hepes (pH 7.6), 1 M NaCl, 1 mM ethylene-bis-(oxyethylenenitrilo)tetraacetic acid (EGTA), 1 mM MgCl2, 0.1% Tween-20, 0.5 mM dithiothreitol (DTT), 10% glycerol) containing protease inhibitors (1 mM phenylmethylsulfonylfluoride (PMSF), 1 μg/ml antipain, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 μg/ml aprotinin) and diluted with the same buffer to the final concentration of about 30 mg total protein per ml. Samples (0.6 ml) were mixed with 10 μg of anti-HA antibody (BAbCO) or 4 μg of anti-green fluorescent protein (GFP) antibody (Roche Diagnostics Corporation), and the mixture was incubated at 4°C overnight. Fifty μl of Protein A-Sepharose beads (Pharmacia) was then added, incubated at 4°C for 1 hour, and washed three times with a mixture of 70% Lysis buffer and 30% RIPA buffer (0.1 M Tris-HCl (pH 7.5), 0.2 M NaCl, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS) and washed five times with a mixture of 70% kinase buffer (50 mM Hepes (pH 7.6), 1 M NaCl, 1 mM EGTA, 2 mM MgCl2, 0.1% Tween-20, 10% glycerol) and 30% RIPA buffer. After final wash, half of the beads were boiled with 20 μl of sample buffer (0.0125 mM Tris-HCl (pH6.8), 2% glycerol, 0.4% SDS, 1% β-mercaptoethanol, 0.0002% bromophenol blue) for 5 minutes and 10 μl of supernatant was separated by 7.5% SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western blotting analysis using anti-HA antibody (BAbCO). To the other half of the beads, 20 μl of assay buffer (kinase buffer containing 0.25 mM ATP, 0.1 mCi/ml [γ32P]ATP, 1 mM DTT, 2 μg/ml antipain, 2 μg/ml leupeptin, 2 μg/ml pepstatin A and 2 μg/ml aprotinin) was added. The reaction mixtures were mixed gently and incubated at 30°C for 30 minutes, with gentle mixing every 10 minutes. The reaction was stopped by the addition of 10 μl of 5×sample buffer and was boiled for 5 minutes, and then 15 μl was loaded onto each lane of a 7.5% SDS-polyacrylamide gel. Incorporation of 32P into Gin4 was visualized and quantified using a Bio imaging analyzer BAS1000 MacBAS (Fujix).
Septins are known to have pleiotropic functions in cytokinesis, bud growth pattern and bud site selection. In our previous work, we demonstrated that Nis1 interacts with septins by two-hybrid assay using Shs1 as bait (Iwase and Toh-e, 2001). During our attempt on functional analysis of Nis1, we performed a two-hybrid screening to find the Nis1 binding proteins. Because septins are bound to Nis1 at its C-terminal region, we used the LexA DBD fusion with C-terminally truncated Nis1 as bait to avoid repeated isolation of septin genes. The AD fusion library was introduced into J21 cells carrying pM-68. Among 1.3×105 transformants, 244 positive clones (His+ and LacZ+) were obtained. DNA sequencing at the junction of each of the 37 randomly selected cloned DNAs revealed that they were classified into seven different ORFs. Among them, 25 clones were found to contain the NAP1 gene that encodes the nucleosome assembly protein (Fujii-Nakata et al., 1992). Nap1 is a member of the mitotic signaling network that regulates Swe1 kinase negatively (Altman and Kellogg, 1997). Seven clones were found to contain the YBR267w gene that encodes protein of an unknown function and contains three C2H2-type zinc finger motifs (Fig. 1B). For the reason discused later, we designated YBR267w as REI1 (required for isotropic bud growth). We were interested in the fact that REI1 interacted with Nis1 that interacted with septins and Nap1, these latter been the members of the mitotic signaling network, and performed further analysis of REI1. Each of the rest of five clones represented a unique ORF and was not analyzed further.
To analyze the function of REI1, we constructed a disruptant of REI1. In the background of W303 and YPH, Δrei1 cells exhibited cold-sensitivity at 15°C and 25°C (Fig. 1A). Δrei1 cells showed a weak defect of cell separation irrespective of growing temperature (data not shown). We did not detect a morphological defect in the disruptant of REI1 (Fig. 2). Septin-GFP was normally localized at the bud neck in the Δrei1 cells (data not shown). There is a homologue of the REI1 gene in the S. cerevisiae genome. The Rei1 homologue, Ylr387c, is a protein of unknown function, and has three dispersed C2H2-type zinc fingers (Fig. 1B). We designated YLR387c as REH1 (REI1 Homologue). Δreh1 cells exhibited no phenotypic change (data not shown). But, double disruptant Δrei1Δreh1 cells exhibited a severe defect in growth (Fig. 1C). The double disruptant cells grew very slowly at 30°C but did not form a colony at 37°C. Double disruptant cells expressing CDC11-GFP grown at 30°C were observed under microscope and a typical image is shown in Fig. 1D. Cells with multiple buds were frequent under this condition while when shifted to 37°C, cells were enlarged. Cold sensitivity of Δrei1 cells was partially suppressed by introducing multicopy plasmid carrying REH1 (Fig. 1E). Although Rei1 was originally isolated on the basis of interaction with Nis1, we found no genetic interactions between NIS1 and either REI1 or REH1.
 View Details | Fig. 1. Genetic characterization of the REI1 gene. (A) Phenotype of the Δrei1 mutant with the W303 background. One of the alleles of REI1 in W303a/α was replaced with the Δrei1::TRP1 gene, and the resulting heterozygous diploid cells (Masa72) were sporulated and dissected. Two wild type and two disruptants were chosen from among the segregants and streaked across YPD plates, each of which was incubated at 15°C for 5 days, at 25°C or 37°C for 3 days. (B) The amino acid sequence alignment of Rei1 and Reh1. Black boxes are identical amino acids. Gray boxes are similar amino acids. Under asterisks are C2-H2 zinc finger regions. (C) Tetrad analysis of diploid cells (Masa77) prepared by crossing Δrei1 cells (Masa47) to Δreh1 cells (Masa49). Tetrads were dissected and grown on YPD plates at 25°C for 5 days. Genotypes of the segregants are indicated on the right. (D) Morphology of Δrei1Δreh1 cells. Cdc11-GFP (pM-3) introduced into Δrei1Δreh1 cells (Masa51). Cells were cultivated at 30°C or 37°C to mid logarithmic phase, and then harvested and observed. The bar indicates 10 μm. (E) Partial suppression of Δrei1 by multicopy of REH1. YCp-REI1 (pM-78), YEp-REI1 (pM-79), YEp-REH1 (pM-85) or vector plasmids (pTS910EU) was introduced into Δrei1 cells (Masa47), and the resulting transformants were streaked on SC-URA plates and incubated at 18°C for 6 days or at 37°C for 3 days. |
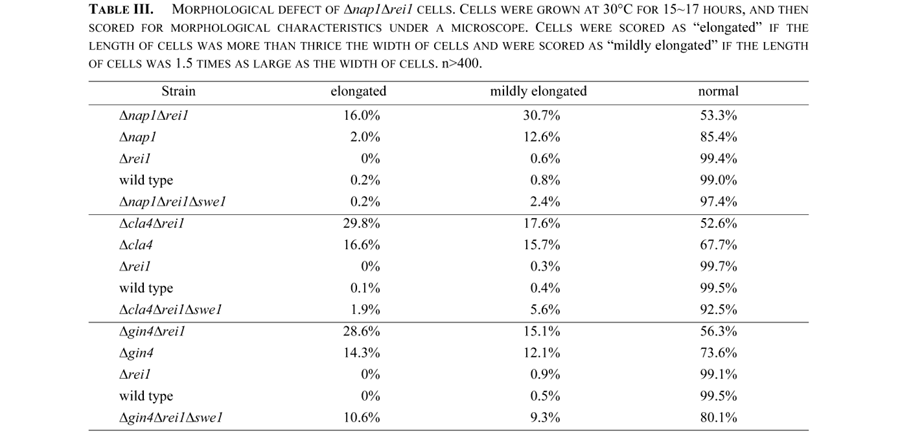
Among Nis1 interacting proteins, Nap1 is known as a member of the mitotic signaling network. Δrei1Δnap1 cells produced an elongated bud more frequently than single disruptant cells (Fig. 2A, Table III). This phenotype was suppressed by a deletion of the SWE1 gene (Fig. 2A bottom, Table III). The elongated bud phenotype in Δgin4, Δnap1, and Δcla4 single disruptant cells was also suppressed by a deletion of the SWE1 gene (Longtine et al., 2000; our unpublished result). These results strongly suggested that Rei1 belonged to the mitotic signaling network. We then extended the double mutant analysis to other members of the mitotic signaling network. The following experiments were carried out at 30°C to avoid growth defect of Δrei1 cells at a lower temperature. The Δrei1Δcla4 and Δrei1Δgin4 strains also exhibited an elongated bud phenotype, which was suppressed by a deletion of the SWE1 gene (Fig. 2B and 2C, Table III). Moreover, Δrei1Δhsl1 cells exhibited a synthetic growth defect (Fig. 3A) and a synthetic morphological defect (data not shown). A triple disruptant Δgin4Δcla4Δrei1 cell was lethal (Fig. 3B). These results support the idea that Rei1 is a member of the mitotic signaling network genes that negatively regulate Swe1 kinase and is required for proper bud growth. A triple disruptant Δrei1Δreh1Δnap1 did not exacerbate the elongated bud phenotype (data not shown). The same results were obtained with Δrei1Δreh1Δcla4 cells and Δrei1Δreh1Δgin4 cells (data not shown). In contrast to Δrei1Δnap1 cells, Δreh1Δnap1 cells did not cause elongated bud morphology (data not shown). These results indicate that REH1, although structurally and functionally similar to REI1, has a distinct function other than bud growth.
 View Details | Fig. 2. Δrei1Δnap1, Δrei1Δcla4 and Δrei1Δgin4 cells displayed an elongated bud morphology that was suppressed by Δswe1. (A) Δrei1Δnap1 cells (Masa54), Δnap1 cells (Masa52), Δrei1 cells (Masa47), wild type cells (W303a) and Δrei1Δnap1Δswe1 cells (Masa55) were grown at 30°C for 15 hours, and cells were then harvested and observed. The bar indicates 10 μm. (B) Δrei1Δcla4 cells (Masa57), Δcla4 cells (Masa56), Δrei1 cells (Masa47), wild type cells (W303a) and Δrei1Δcla4Δswe1 cells (Masa58) were grown at 30°C for 17 hours, and then cells were harvested and observed. The bar indicates 10 μm. (C) Δrei1Δgin4 cells (Masa60), Δgin4 cells (Masa59), Δrei1 cells (Masa47), wild type cells (W303a) and Δrei1Δgin4Δswe1 cells (Masa61) were grown at 30°C for 16 hours, and cells were then harvested and observed. The bar indicates 10 μm. |
 View Details | Fig. 3. Synthetic phenotype of Δrei1 and Δhsl1 or Δgin4Δcla4. (A) The Δrei1Δhsl1 strain is lethal. Tetrad analysis of diploid cells (Masa82) from a cross between Δrei1 cells (Masa47) and Δhsl1 cells (Masa84) is shown. Tetrads were dissected and grown on YPD plate at 25°C for 5 days. Genotypes of the segregants are indicated on the right. (B) The Δgin4Δcla4Δrei1 strain is lethal. Tetrad analysis of diploid cells (Masa83) from a cross between Δrei1Δgin4 cells (Masa60) and Δrei1Δcla4 (Masa57) is shown. Tetrads were dissected and grown on YPD plates at 30°C for 5 days. The genotypes of the segregants are indicated on the right. |
GIN4, NAP1, CLA4, ELM1 and septin genes are known as the genes that are required for isotropic bud growth at the upstream of the SWE1 function. These genes were reported to be required for Gin4 hyperphosphorylation or Gin4 kinase activity (Altman and Kellogg, 1997; Sreenivasan and Kellogg, 1999; Tjandra et al., 1998; Carroll et al., 1998). In Gin4 kinase assay using Histone H1 as substrate, a band of phosphorylated protein of the same as Gin4 was seen. Phosphorylation of this protein is not detected in a deletion or a mutant of GIN4 cells, suggesting that it is the Gin4 protein itself and that Gin4 is capable of undergoing autophosphorylation (Altman and Kellogg, 1997). The fact that Gin4 phosphorylation occurred in parallel with the appearance of Gin4-associated kinase activity is consistent with the idea that Gin4 is autophosphorylated (Altman and Kellogg, 1997). We assayed Gin4 kinase activity using immunoprecipitated Gin4-2xHA or Gin4-GFP and γ-32P-ATP. As a result, we detected phosphorylation signals at the position corresponding to those of Gin4-2xHA and Gin4-GFP bands, confirming that Gin4 undergoes autophosphorylation (Fig. 4A).
 View Details | Fig. 4. Assay of Gin4 kinase. Tagged Gin4 was immunoprecipitated with anti-tag antibody and the immunoprecipitate was used as an enzyme source for kinase assay. (A) Gin4 is autophosphorylated. lane 1, Gin4-2xHA (pM-86) in Δgin4 cells (Masa59); lane 2, or Gin4-GFP (pM-87) in Δgin4 cells (Masa59); lane 3, the vector plasmid (pTS903CU) in wild type cells (W303a); lane 4, the vector plasmid (pTS910CU) in wild type cells (W303a). (B) Deletion of REI1 recovered Gin4 kinase activity in the Δnap1 cells to the wild type level. Lane 1, wild type cells (Masa59 [pM-86]); lane 2, Δrei1 cells (Masa60 [pM-86]); lane 3, Δnap1 cells (Masa62 [pM-86]); lane 4, Δnap1Δrei1 cells (Masa63 [pM-86]); lane 5, control cells (W303a [pTS903CU]). Using NIH Image ver. 1.62, the background counts of each lane were subtracted from the Gin4 kinase activity counts of each lane. The value of lane 5 was subtracted from that of lanes 1–4. The value of wild type cells was expressed as 1 and the kinase activities of other strains were expressed as relative values to that of wild type cells. Each bar indicates an average of four kinase assays. (C) Estimation of Gin4 protein. Electrophoresis was carried out as described above. Gin4 was detected by Western blotting using anti-HA antibody and Cdc28 by anti-PSTAIRE antibody (Santa Cruz Biotechnology). Using NIH Image ver. 1.62, background counts of the each lane were subtracted from the Gin4 protein counts of each lane. The counts of lane 5 was subtracted from that of lane 1–4. Each value was divided by the counts of Cdc28 protein as an internal reference. The value of wild type cells was expressed as 1 and the values of other strains were expressed as relative values to that of wild type cells. |
To assign the Rei1 function in the mitotic signaling network, we examined whether Rei1 affects Gin4 kinase or not. We assayed Gin4 kinase activity using Gin4-2xHA as substrate in Δrei1 cells and in Δnap1Δrei1 cells. The same level of Gin4 kinase activity was observed in Δrei1 cells that show normal bud growth and in wild type cells (Fig. 4B, lane 1 and lane 2). On the other hand, Gin4 kinase activity of Δnap1 cells was low as shown by Altman and Kellogg (1997) (Fig. 4B, lane 3). An amount of Gin4 protein in Δnap1 cells was also lower than that in wild type cells, indicating that the lower activity of Gin4 kinase in Δnap1 cells is due to the lower level of Gin4 protein (Fig. 4C). Interestingly, the level of Gin4 protein, and Gin4 kinase activity as well, was recovered nearly to the wild type level by introducing the Δrei1 mutation into the Δnap1 cells (Fig. 4B and C lane 4). It should be noted that the bud of Δnap1Δrei1 cells was more elongated than that of Δnap1 cells. On the other hand, the level of Gin4 kinase activity of Δnap1Δreh1 cells was the same as that of Δnap1 cells, indicating that REH1 does not affect Gin4 kinase (data not shown). These results imply that Rei1 functions to keep Gin4 activity low in Δnap1 cells, and that Gin4 kinase activity is not required for isotropic bud growth. To examine the effect of Rei1 on Gin4 kinase activity, Gin4 kinase activity was assayed using Gin4-2xHA immunoprecipitate prepared from the Δnap1Δrei1 cells as enzyme source in the presence of purified T7-Rei1-6xHis expressed in E. coli. Protein sample (1 μg) was analyzed by 10% SDS-PAGE followed by CBB staining (Fig. 5A right panel). T7-Rei1-6xHis inhibited the Gin4 kinase activity in a dose-dependent manner (Fig. 5A), whereas bovine serum albumin (BSA) as a control had no effect on the Gin4 kinase activity (Fig. 5B). At present, the possibility that the tag sequence (T7-6xHis) can inhibit Gin4 kinase activity remains to be eliminated.
 View Details | Fig. 5. Gin4 kinase activity is inhibited by Rei1 in vitro. (A) Left panel: Gin4 kinase was assayed using Gin4-2xHA immunoprecipitated from Masa59 [pM-86] cells as an enzyme source with an indicated amount of T7-Rei1-6xHis protein. The indicated amount of T7-Rei1-6xHis protein was separated by SDS-PAGE and stained with Coomassie brilliant blue. Using NIH Image ver. 1.62, the background counts were subtracted from the Gin4 kinase counts. Gin4 kinase assayed without T7-Rei1-6xHis protein was expressed as 1 and kinase activities under other conditions were expressed as relative values. Right panel: 1 μg T7-Rei1-6xHis protein was analyzed by 10% SDS-PAGE followed by CBB staining. (B) Gin4 kinase activity was not inhibited by bovine serum albumin (BSA). Gin4 kinase was assayed as above with BSA (Pierce) in place of T7-Rei1-6xHis protein. The indicated amount of BSA was separated by SDS-PAGE and stained with Coomassie brilliant blue. Gin4 kinase activity was normalized as described in (A). |
The result of our Gin4 kinase assay is not consistent with the presently accepted interpretation of the regulation of bud morphology in that the loss of Gin4 kinase activity causes an elongated bud morphology. Here we assume that Gin4 kinase activity may not be required for isotropic bud growth. If this is the case, then the morphological defect caused by disrupting the GIN4 gene should be suppressed by a kinase inactive Gin4 mutant gene. We constructed a plasmid carrying the kinase inactive gin4K48A gene or that carrying the gin4ΔKD gene missing the kinase domain, and each plasmid, as well as the plasmid carrying the wild type GIN4 as reference, was introduced into Δgin4Δrei1 cells which exhibited elongated bud morphology. GIN4, gin4K48A or gin4ΔKD suppressed the elongated bud phenotype, but the vector plasmid did not (Fig. 6). The same result was obtained by introducing the plasmid into Δgin4 cells (data not shown). This is a clear indication that the kinase activity of Gin4 is not needed for isotropic bud growth.
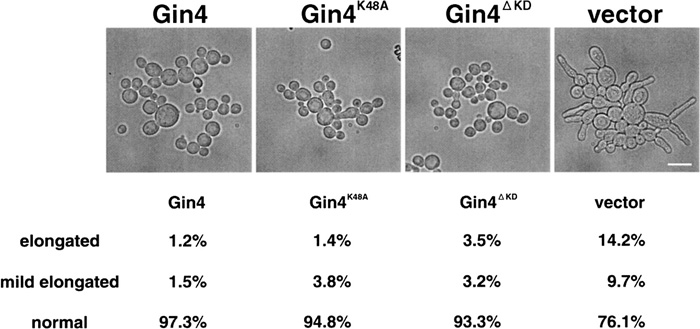 View Details | Fig. 6. gin4K48A and gin4ΔKD suppressed a morphological defect of Δgin4Δrei1 cells. Δgin4Δrei1 cells (Masa60) were introduced by wild type GIN4 plasmid (pM-86), GIN4K48A plasmid (pM-90), GIN4ΔKD plasmid (pM-103) or vector plasmid (pTS903CU). Cells were grown at 30°C for 15 hours in SC-URA medium, and then cell morphology was examined under microscope. The bar indicates 10 μm. |
Proteins belonging to the mitotic signaling network are known to be localized to the bud neck; they are septins (Cdc3, Cdc10, Cdc11, Cdc12 and Shs1), Elm1, Hsl7, Gin4, Hsl1, Kcc4, Swe1 and Clb2 (Moriya and Isono, 1999; Shulewitz, 1999; Longtine et al., 2000; Hood et al., 2001). To determine the localization of Rei1, we constructed a GFP fusion Rei1. Because the signal from a single copy REI1-GFP fusion gene was not detected, we observed a signal from a multicopy REI1-GFP fusion or a single copy REI1-8xGFP fusion gene. These plasmids (carrying single copy or multicopy REI1-GFP fusions, and single copy REI1-8xGFP fusion gene) suppressed the cold sensitivity of the Δrei1 cells, indicating that these Rei1-GFP fusions are all functional (Fig. 1E). Rei1-GFP was localized to the cytoplasm (Fig. 7A, data of Rei1-8xGFP fusion are not shown) at 25°C. When the culture was shifted to 37°C, Rei1-GFP aggregated into several discrete masses, which were re-dispersed to the cytoplasm by returning the culture to 25°C (Fig. 7B).
 View Details | Fig. 7. Localization of Rei1. (A) Δrei1 cells (Masa47) containing multicopy plasmids of Rei1-GFP (pM-79) were grown at 25°C to mid-logarithmic phase and fixed. After fixation, cells were harvested and were stained by 4',6-diamidino-2-phenylindole (DAPI). (B) Masa47 (pM-79) cells were grown to logarithmic phase at 25°C. A part of the culture was shifted to 37°C. At 10 minutes and 30 minutes after the shift, GFP was observed as above. After 30 minutes incubation at 37°C, the culture was shifted to 25°C. After 2.4 hours, GFP was observed. Bars indicate 10 μm. |
Kellogg et al. (1995) showed that Nap1 is primarily a cytoplasmic protein by immunofluorescence experiments. We constructed a GFP fusion Nap1, and observed a Nap1-GFP signal. As expected, the Nap1-GFP signal was detected in the cytoplasm. Nap1-GFP was also observed at the incipient bud site and at the bud neck as a double ring during cell cycle (Fig. 8A). Many of the members of the mitotic signaling network were localized to the bud neck in a septin dependent manner (Bouquin et al., 2000; Shulewitz, 1999; Longtine et al., 2000; Longtine et al., 1998; Barral et al., 1999; Hood et al., 2001). To examine whether the bud neck localization of Nap1-GFP depends on septins or not, we observed Nap1-GFP in septin mutants. In either the cdc3-1 strain or the Δcdc10 strain, Nap1-GFP was not detected at the bud neck but was localzed at the bud tip at a restrictive temperature (Fig. 8Ba–d). Adiditionally, in elm1 mutant cells, Nap1-GFP was observed at the bud neck and often at the bud tip (Fig. 8Be).
 View Details | Fig. 8. Localization of Nap1. (A) NAP1-GFP plasmid (pM-65) introduced into Δnap1 cells (Masa52, a–e). No tagged NAP1 plasmid (pM-66) was introduced into Δnap1 cells (Masa52, f). Cells were cultivated at 25°C to mid-logarithmic phase and harvested, and then GFP was observed. The bar indicates 10 μm. (B) Nap1-GFP plasmid (pM-65) introduced into cdc3-1Δnap1 cells (Masa64, a; 25°C, b; 37°C), Δcdc10Δnap1 (Masa65, c; 25°C, d; 37°C) cells and elm1Δnap1 cells (Masa66, e; 25°C). Each culture was grown at 25°C to mid-logarithmic phase and a part of it was shifted to 37°C. After 3.0 hours of incubation at 37°C, the cells were harvested, and GFP was observed. The bar indicates 10 μm. |
As summarized in Table IV, various multiple mutants of mitotic signaling network genes displayed an elongated cell morphology and some showed a growth defect in addition to a morphological defect. Introduction of the Δswe1 mutation in these strains cured the morphological abnormality but not the growth defect. Swe1 is an equivalent of the Wee1 kinase of S. pombe, which inhibits CDK by phosphorylating tyrosine 19 of Cdc28. Dephosphorylation of this phospho-tyrosine is crucial for the activation of the Cdc28/Clb2 kinase. In contrast to the fact that this dephosphorylation is essential for cell cycle progression in the fission yeast, phosphorylation/dephosphorylation of Cdc28 at this tyrosine of S. cerevisiae is not critical for growth because of the presence of redundant cyclin partners in the budding yeast. Barral et al. (1999) demonstrated that the temperature-sensitive mutant cdc12-1, when shifted to 37°C, produced an elongated bud. This elongated bud phenotype was suppressed by Δswe1 but the temperature-sensitive growth was not. The cdc12-1Δswe1 strain showed a severer growth defect than the cdc12-1 strain and this double mutant lost viability faster than the cdc12-1 strain at 37°C. These genetic interactions between cdc12-1 and Δswe1 indicate that the mitotic signaling network is not a single pathway.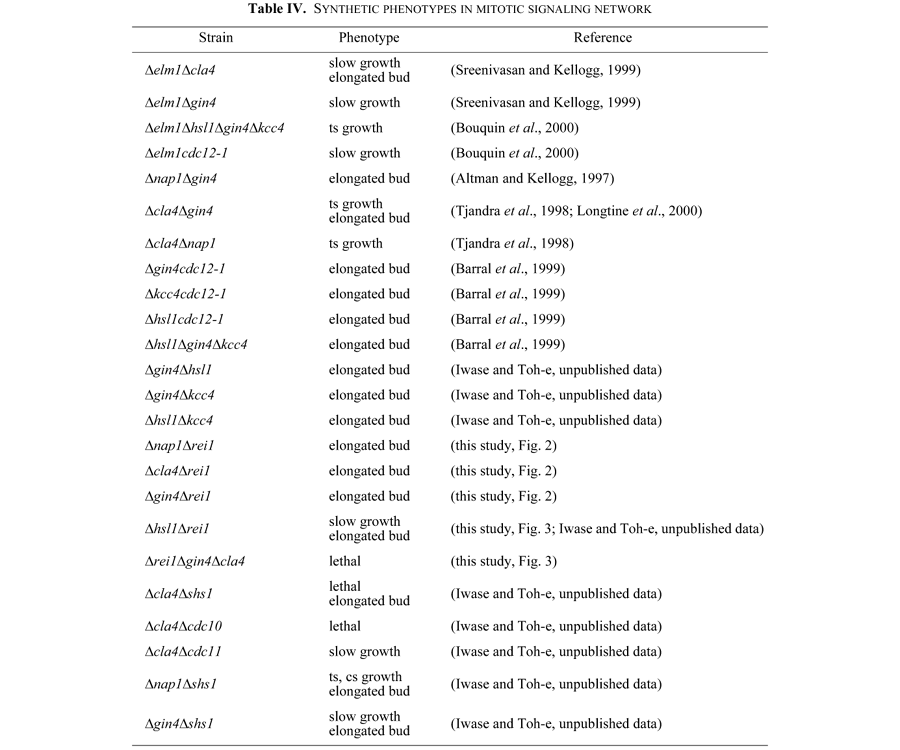
When the GIN4 derivative without kinase activity was introduced into the Δrei1Δgin4 strain, the elongated morphology was cured, indicating that Gin4 kinase activity is not necessary for changing the bud growth pattern (Fig. 6). This conclusion is in conflict with that proposed previously. Longtine et al. (1998) showed that the Gin4 kinase domain interacted with the C-terminal portion of Cdc3 septin in a two-hybrid assay and mild overexpression of GIN4 could rescue both the viability and the morphology of a cdc12 temperature-sensitive mutant at a semi-restrictive temperature. But, introduction of the kinase dead mutant gene, GIN4K48M or GIN4K48A , did not rescue temperature-sensitivity of the cdc12 mutant, a morphological defect of the cdc12 mutant at intermediate temperatures or the synthetic lethality of the Δgin4cdc12 double mutant, and cells expressing only a kinase-dead allele Gin4K48M displayed conspicuously abnormal septin organization (Longtine et al., 1998). These results indicate that Gin4 kinase activity is required for the proper septin organization or assembly. However, the observation that a kinase-dead version of Gin4 did not suppress temperature-sensitivity and the morphological defect of the cdc12-6 mutant, does not necessarily indicate that the kinase activity is required for isotropic bud growth. It is still possible that Gin4 kinase activity is required for Gin4 to interact with cdc12-6 mutant protein to recover the septin structure and growth ability, which automatically results in isotropic bud growth. It is thus preferable to use mutants exhibiting only the morphological defect to assess Gin4 kinase function in the determination of the bud growth pattern as we described in this work. Our results indicate that Gin4 has at least two functions; one is a kinase dependent function that is probably involved in the septin-organization, and the other is a kinase independent function that is involved in the bud growth in the mitotic signaling network. In contrast to Gin4, kinase activity of another Nim1-related kinase, Hsl1, is needed for isotropic bud growth (our unpublished result).
Also Rei1 and Nap1 each has at least two functions associated with two different Gin4 functions. Taken together, there are two functional Gin4 branches based on two Gin4 functions; one is septin organization exerted by Gin4 kinase activity and the other is control of bud morphology exerted by non-kinase activity of Gin4.
REI1, a non-essential gene, was isolated by two-hybrid screening using NIS1 as bait. There is one closely related gene of REI1 in the yeast genome and we have designated this gene as REH1. When both genes were disrupted, cells grew slowly at 25°C and showed temperature-sensitive growth. This synthetic effect, along with the fact that the high-copy of REH1 suppressed a defect of Δrei1, indicates that these two genes have some overlapping functions; however, REH1 was found to have no function in the control of bud morphology. Δrei1 exhibited a synthetic effect with some mutants of the mitotic signaling network genes such as Δnap1 and Nim1-related kinase mutants. The double mutants showed an elongated bud phenotype, which was suppressed by the Δswe1 mutation (Fig. 2). For these reasons, REI1 is likely to belong to the mitotic signaling network although the exact function of Rei1 remains unknown. In this study, we found a clue to the biochemical function of Rei1 as follows. We found that Gin4 protein and its kinase activity in the Δnap1Δrei1 cells recovered nearly to the wild type level (Fig. 4). From these results, we inferred that Rei1 may have a negative effect on Gin4 protein in the absence of Nap1, and we found that this is likely the case (Fig. 5). Rei1 inhibited Gin4 kinase activity in vitro, although its inhibitory activity was very weak. This result suggests that there is some interaction between Rei1 and Gin4 at the protein level. However, the significance of inhibition of Gin4 kinase activity by Rei1 in the mitotic signaling network remains obscure.
In contrast to the fact that all the members of the mitotic signaling network so far tested are localized to the bud neck, at least at some stage of the cell cycle, Rei1 is always distributed in the cytoplasm (Fig. 7). Thus, Rei1 is a unique protein among the mitotic signaling network proteins. However, the cytoplasmic localization of Rei1 does not exclude the possibility that Rei1 can interact with proteins at the bud neck.
Elongated bud morphology due to a defect in a mitotic signaling network gene is suppressed by Δswe1, indicating that Swe1 is a key target of the mitotic signaling pathway. The data summarized in Table IV strongly support a model that Swe1 kinase activity is controlled by multiple parallel pathways as proposed by Longtine et al. (2000). Our observations that double mutants containing Δrei1 and one member of the mitotic signaling network displayed an elongated bud phenotype and that this defect was suppressed by Δswe1, indicate that Rei1 is one module of the Swe1 regulators.
The authors would like to express their thanks to Y. Kikuchi, Y. Matsui, J. Takeuchi, and T. Michimoto for yeast strains and plasmids. MI was a recipient of a grant from JSPS for pre-doctoral fellows.
|