| To whom correspondence should be addressed: Ritsu Kamiya, Department of Biological Sciences, Graduate School of Science, University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. Tel: +81–3–5841–4426 Abbreviations: FRAP, fluorescence recovery after photobleaching; IFT, intra-flagellar transport; GFP, green fluorescent protein; TAP, Tris-acetate-phosphate. |
Cilia and flagella of eukaryotes are cell organelles that produce bending waves and cause cellular swimming or water flow on the cell surface. In mammals, these organelles are used in a variety of functions such as swimming of sperm, clearance of airways, transport of oocytes, and determination of the left-right asymmetry in embryos (for review, see Ibanez-Tallon et al., 2003). Cilia and flagella contain in their cores a highly complex yet regular structure called an axoneme, consisting of nine outer-doublet microtubules surrounding a pair of singlet microtubules and various associated structures. This structure, known as the 9+2 structure, is composed of more than 250 proteins and, strikingly, is conserved in most organisms ranging from protozoa to humans. How such a complex structure is formed and maintained is a major problem in cell biology (Gibbons, 1981; Silflow and Lefebvre, 2001).
Recent studies with Chlamydomonas have shown that flagella are equipped with a special transport system, called the intra-flagellar transport (IFT), for conveying flagellar proteins along the length of the axoneme (Kozminski et al., 1993; Rosenbaum and Witman, 2002; Cole, 2003). The IFT consists of two oppositely directed transport systems: the anterograde transport that carries proteins in the base to tip direction driven by kinesin-II (Kozminski et al., 1995; Cole et al., 1998), and the retrograde transport that operates in the tip to base direction driven by a special type of cytoplasmic dynein (Pazour et al., 1999; Porter et al., 1999). Flagella do not grow in mutants lacking either transport system, indicating that IFT is essential for flagellar formation. Both the anterograde and retrograde transport systems appear to constantly operate even in fully grown flagella. This suggests that flagella are dynamic structures in which protein components are being incessantly supplied or removed by IFT.
In accordance with the possible dynamic nature of ciliary or flagellar structure, recent experiments have suggested that the axonemal proteins are being dynamically turned over in stationary cilia and flagella. For example, injection of radioactive amino acid into ciliated embryos of sea urchin resulted in the labeling of some axonemal proteins independently of ciliary growth, suggesting that those proteins are being constantly renewed (Stephens, 1997). Similar conclusions were reached in experiments using Chlamydomonas grown in radioactive media (Song and Dentler, 2001). These experiments, however, have not directly shown how a particular protein is exchanged in a single cilium or flagellum. Thus, whether a given protein can be exchanged over the entire length of cilia and flagella, or only at their tips has yet to be clarified. Nor is it certain whether or not the exchange is carried out in a treadmilling manner, such that new proteins are incorporated at the base (or tip) while old proteins are removed from the tip (or base). To elucidate these problems, we need to visualize the turnover in cilia and flagella in live cells.
In this study, we thus attempted to directly observe the turnover of an axonemal protein, actin, in live Chlamydomonas cells, using a fluorescence-recovery-after-photobleaching (FRAP) method. Chlamydomonas is an organism that offers a variety of flagella-deficient mutants and has been most extensively used in studies on flagellar assembly and motility. Actin is a flagellar protein existing mostly as a subunit of inner arm dyneins (Piperno and Luck, 1979). The FRAP method has been used for detection of protein turnover in a variety of biological systems, including membranes and cytoskeletons, although, to our knowledge, no studies have been carried out to examine the protein dynamics in the axoneme. In most FRAP experiments, the protein in question is first fluorescently labeled and incorporated into the cellular structure by microinjection or by expression of conjugate protein with green-fluorescent protein (GFP), followed by photobleaching of a small area by irradiation with strong light. A problem in applying FRAP techniques to Chlamydomonas has been that it is difficult to introduce fluorescent proteins, or to express GFP-conjugated proteins. Recently, however, our laboratory has developed a method to introduce exogenous proteins into live Chlamydomonas cells using electroporation, and has shown that fluorescently labeled actin is functionally incorporated into inner arm dyneins in a mutant lacking the conventional actin gene (Hayashi et al., 2001). Using this technique, we carried out the first attempt to examine the dynamic exchange of actin in Chlamydomonas flagella.
A wild type Chlamydomonas reinhardtii, 137C, and a double mutant, ida5oda1, were used. The mutant ida5 lacks the gene of conventional actin and also lacks a subset of inner arm dyneins that contain actin as subunits (Kato-Minoura et al., 1997). The mutant oda1 entirely lacks outer arm dynein (Kamiya, 1988). These mutants display slow swimming, while the double mutant is non-motile. The cell was grown in Tris-acetate-phosphate (TAP) media on a 12 h/12 h, light/dark cycle, with aeration.
Flagella and axonemes were prepared by the method of Witman et al. (1978). SDS-PAGE and Western blot analysis of the isolated axonemes were carried out after Laemmli (1970) and Towbin et al. (1979), respectively. Anti-actin antibody was described elsewhere (Sugase et al., 1996).
Rabbit skeletal muscle actin was prepared by the method of Pardee and Spudich (1982). Fluorescently-labeled actin was produced by labeling actin with tetramethylrhodamine-5-maleimide (Molecular Probes, Eugene, OR) according to the method of Tait and Frieden (1982). Briefly, actin was polymerized in polymerization solution (10 mM HEPES (pH 7.0), 100 mM KCl, 10 mM MgSO4, 0.2 mM ATP, 0.1 mM CaCl2), mixed with 5-fold molar excess of dye, and allowed to react for 2 h at room temperature. The addition of DTT stopped the reaction. Free dye molecules were removed by gel filtration using a Sephadex G-25 column. The molar ratio of the bound dye to actin was ~1.0. This procedure has been shown to produce covalent modification of the Cys-374 residue of actin (Tait and Frieden, 1982).
Fluorescent monomeric actin was introduced into ida5oda1 cells using electroporation, following the method of Hayashi et al. (2001). Briefly, cells were treated with autolysin to remove the cell walls, and subjected to electroporation in the presence of 0.1 mg/ml labeled actin, 0.2 mM ATP-imidazole (pH 7.4), 0.1 mM CaCl2, 0.5 mM 2-mercaptoethanol, and 60 mM sucrose. The final cell density was adjusted to 5×107/ml. An ECM600 electroporation apparatus (BTX, Holliston, MA) was used. Electric pulses of variable voltage were applied, with the resistance set to 24 ohms and the conductance to 1200 μF. Flagella were shed upon application of an electric pulse, but re-grew to full length in about 3 hours. Actin-incorporated cells displayed flagellar motility and fluorescence. The amount of actin introduced was apparently sufficient for the cell to recover all actin-containing dyneins, since the ida5oda1 cells that became motile after electroporation displayed swimming velocities indistinguishable from those of oda1 cells (Hayashi et al., 2001). Electroporated cells were kept overnight under dark conditions until used in photobleach experiments.
Cells that had regained normal flagellar lengths after actin introduction were observed with an Olympus IX70 inverted fluorescence microscope, equipped with a VS4-1845 image intensifier (Videoscope International, Dulles, VA) and a chilled CCD video camera (C5985, Hamamatsu Photonics, Hamamatsu, Japan). The excitation light for fluorescence microscopy was passed through a 6% ND filter and a short-pass filter (<610 nm) to minimize bleaching due to observation and to lessen the effect of chloroplast autofluorescence, respectively. Actin fluorescence in a short (~1 μm) segment of a flagellum was bleached by irradiation with a 20 mW green laser (Uniphase Co. Oxon, UK), delivered through the 100X objective lens used for observation, and focused on the specimen. The position to be photobleached was set at 50–70% of the flagellar length from the base. The intensity of the laser beam was adjusted using ND filters. Laser light for photobleaching was usually irradiated for about 1 sec.
To keep live cells in healthy condition and restrict their movement within a fixed area while being monitored after photobleaching for up to 2 hours, we used a chamber made up of a glass slide coated with a thin layer of agar (containing the TAP medium), a coverslip, and spacers. Cells were sandwiched between the agar layer and the glass surface; this allows some freedom of flagellar movement, a condition that prevents flagellar shrinking. Observation and video-recording of the bleached portion were carried out for 2 seconds at ~15 minute intervals. Acquired images were averaged, stored as still images, and analyzed with an Argus 20 image processor (Hamamatsu Photonics) and the NIH Image software. Image intensities were measured in a 0.5 μm long flagellar segment around the photobleached point, as well as in a 1.5 μm long segment that was apparently not bleached. The ratio of the average pixel intensities in these two segments was taken as the measure of fluorescence recovery.
Actin is a subunit of several species of inner arm dyneins in Chlamydomonas flagella. Previous studies have shown that greater than 70% of flagellar actin is contained in the axoneme, mostly as a dynein subunit (Piperno and Luck, 1979). To confirm the predominant axonemal localization of actin, we first performed Western blot analysis on wild type axoneme to semi-quantitatively compare the amount of actin between the axonemal fraction and the detergent-soluble fraction. Relative band intensities in the immunoblot pattern indicated that 70–80% of the total actin is in fact present in the axoneme fraction (Fig. 1).
 View Details | Fig. 1. Actin localization in Chlamydomonas flagella assessed by immunoblot. Wild-type flagella (Flagella) were demembranated with 0.5% NP40 and separated by centrifugation into a detergent-soluble “membrane and matrix” fraction (M & M) and insoluble axonemal fraction (Axonemes). Those samples were suspended in the same volume of sample buffer, separated by SDS-PAGE, transferred to a membrane, and blotted with anti actin antibody. Left panel shows the SDS-PAGE pattern stained with silver; right panel shows the immunoblot pattern. Arrowhead: band with actin. The actin band intensities in the immunoblot patterns of the axoneme and the M & M fraction relative to the band intensity in the flagella are 71% and 16%, respectively. Therefore, 70–80% of the total actin should be present in the axoneme fraction. |
For photobleaching experiment, we labeled actin with tetramethylrhodamine, which can be photobleached by green laser light, and introduced it into the double mutant ida5oda1. This double mutant is non-motile due to the absence of outer arm dynein and a subset of inner arm dyneins. Upon incorporation of exogenous actin, it became motile due to the recovery of missing inner arm dyneins. As in our previous study (Hayashi et al., 2001), electroporation at ~1200 V/cm was found to be optimal, where about 50% of total cells were killed and 10–20% of surviving cells regained motility. Those motile cells displayed rhodamine fluorescence in their flagella (data not shown). These observations are essentially the same as reported by Hayashi et al. (2001).
We first tried to photobleach a small segment of flagella in cells that were tethered between a glass slide and a coverslip. Those cells adhered to the glass surface by the two flagella pointing in opposite directions. However, we found that the flagella tended to detach or shorten during the observations lasting for ~2 hours. In addition, when cells were loosely tethered, they frequently escaped from the field of observation through the gliding movement exerted by the flagellar membrane. Speculating that these phenomena might have been caused by the excessively strong attachment of the flagella to a solid surface, we coated the glass slide with a thin layer of agar. Cells were pressed between those two surfaces. With this modification, flagellar shrinkage occurred less frequently and cells tended to remain in the same area. This method thus enabled us to monitor fluorescence recovery in flagella of a particular cell for more than 2 hours. Under these conditions, cells occasionally displayed bursts of vigorous beating of photobleached flagella, while remaining in a small area. Although this behavior interfered with the recording of fluorescence images, it at the same time suggested that photobleaching did not cause serious damage to the flagellar motility machinery.
In almost all cases, photobleached areas in the mid portions of flagella were found to increase the fluorescence intensity over a time range of several tens of minutes. Typical records are shown in Fig. 2. Here, the fluorescent intensity was dropped to almost zero just after application of laser light (time 0), but slightly recovered in 17 min. After 35 min, no further recovery became evident. As an estimate of recovery, we took the ratio between the fluorescence intensity averaged around the bleach point to that of the area that apparently was not bleached (Fig. 2B). It demonstrates a clear fluorescence recovery in the initial 30 min. Prolonged observation for more than 2 hours resulted in a decrease in fluorescence of total flagella, causing a large scatter in the data. As evident in these photographs, this experimental system had a problem in that the flagellum, with some freedom of movement, did not always stay in focus. Because of this, the flagellar shape and the intensity profile varied from time to time. This should have caused large errors in the estimate of fluorescence recovery. Despite such difficulties, however, it seems clear that the bleached area underwent significant recovery without changing its location from the flagellar base during the 2.5 hours of observation.
 View Details | Fig. 2. Fluorescence recovery after photobleaching in a flagellum that has incorporated fluorescent actin. A, a series of fluorescence micrographs with intensity profiles (graphs shown at the left side). The number at the upper right corner in each photo indicates the time after photobleaching; the first photo without a number is an image taken before photobleaching, and the photo with time 0 is an image immediately after photobleaching. Arrowheads indicate the position of the photobleached segment. This particular flagellum significantly elongated during observation for unknown reasons. B. Time course of the fluorescence recovery as assessed by the ratio between the image intensities at the bleached portion and non-bleached portion. |
Fig. 3 shows the time course of the fluorescence recovery observed in 14 independent specimens. The speed and extent of recovery greatly differed from a cell to another. Overall, however, the photobleached area appears to recover 10–40% of the initial intensity within 1 hour.
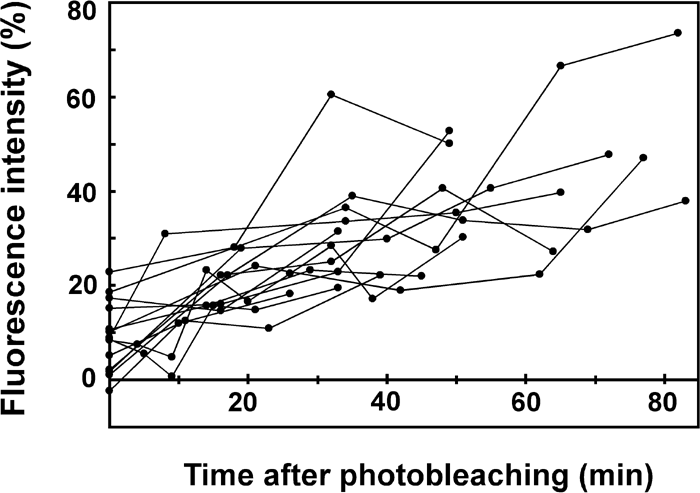 View Details | Fig. 3. Fluorescence recovery in 14 independent samples. The degree of recovery was assessed as in Fig. 2. |
In this study, we introduced fluorescently labeled actin into Chlamydomonas cells, photobleached small portions of actin-loaded flagella, and found that fluorescence recovered to 10–40% of original levels within 1 hour. Because the photobleaching should have destroyed only the attached dye and not the protein itself, this result indicates that 10–40% of actin was replaced by new one during the experiment. As reported previously and confirmed in this study, a large majority of actin is present as an axonemal component, mainly as a subunit of inner arm dyneins. Therefore, the present study has provided the first direct evidence that an axonemal protein undergoes dynamic turnover. The present study, however, cannot give information as to whether inner arm dynein as a whole or actin alone is replaced. We think it is likely that the inner arm dynein as a whole undergoes turnover, since actin has been shown to be essential for the attachment of inner arm dyneins to the outer doublet microtubules. However, this must be confirmed by experiments using other subunits of dynein.
Although almost all samples displayed fluorescence recovery to some extent in the present experiment, the time course and extent of recovery showed a large difference from one specimen to another. Part of the reason for this data scatter must be the difficulty in measuring the fluorescence intensity along the length of the flagellum, which was allowed to move slightly during the experiment and thus did not remain in focus. The flagellar movement was necessary to prevent flagellar shrinkage, and was inevitable even when we used mutants with impaired motility (data not shown). Another reason for this scatter might be that the rate and extent of protein turnover in fact differed from one cell to another, depending on the cell’s condition. For example, the exchange may well be influenced by the freedom of movement of the flagellum. Despite the large data scatter, the time course we estimated qualitatively agrees with the conclusion obtained in previous studies using isotope labeling; for example, Song and Dentler (2001) reported that at least 20% of flagellar proteins are exchanged within a 6-hour period, and that the rate of exchange varies among different proteins.
Several features of the observed fluorescence warrant consideration. First, the fluorescence recovery appeared to plateau at a certain percentage and never reached 100%. Although longer observations may reveal a 100% exchange, it is also possible that only part of actin undergoes turnover; the turnover rate may differ greatly between different species of inner arm dyneins. This possibility must be explored in future studies. Another point is that the position of the bleached portion did not move while its intensity was being recovered. This implies that the microtubules to which actin is attached do not undergo “treadmilling”, such that tubulin continuously attaches to one end and detaches from the other, although tubulin turnover based on such a mechanism has been postulated previously (Stephens, 2000).
A FRAP experiment as performed in this study is a powerful approach to study the dynamics of other axonemal proteins. We chose actin here only because it can be easily prepared in large quantities and because a previous study in our laboratory has established the method for its introduction into live cells. However, we have also succeeded in introducing another light chain of inner arm dynein, p28, by a similar method (Hayashi et al., 2002). Use of this and other dynein light chains will be useful for elucidating the features of protein turnover specific to particular inner arm dynein or outer arm dynein. We could also perform similar experiments using cilia and flagella in other organisms, in particular those that permit expression of GFP-conjugated proteins. Thus the present study must be taken as a starting point for future experiments, which we hope will elucidate the detailed features of the turnover of individual axonemal proteins, as well as the mechanism by which the turnover takes place.
|