| To whom correspondence should be addressed:Yukiko Gotoh, Ph. D., Institute of Molecular and Cellular Biosciences, University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-0032, Japan. Tel: +81–3–5841–8473, Fax: +81–3–5841–8472 Abbreviations: A2A-R, adenosine A2A receptor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; hGH, human growth hormone; LDH, lactate dehydrogenase; NPY, neuropeptide Y; PI3K, phosphatidylinositol-3 kinase; PKI, protein kinase inhibitor; PKA, protein kinase A. |
Adenosine is a potent biological mediator that affects numerous cell types including neural cells, platelets, neutrophils and smooth muscle cells (Daval et al., 1991). Adenosine is released from metabolically active cells by facilitated diffusion or is generated by degradation of released ATP in the extracellular space (Daval et al., 1991). At present, four adenosine receptor subtypes have been identified: A1, A2A, A2B and A3. They are all G protein-coupled receptors (GPCR) coupling to classical second messenger pathways such as modulation of cAMP production or the phospholipase C pathway (Klinger et al., 2002; Olah and Stiles, 1995).
In the central nervous system, A2A-R is highly expressed in corpus striatum, nucleus accumbens and olfactory tubercles and colocalizes with the D2 dopamine receptor in GABAergic striopallidal neurons (Ferre et al., 1993). A2A-R is also expressed at low levels in other areas of the brain (Rosin et al., 1998). A2A-R has been regarded as a potential therapeutic target in protecting against neurodegenerative disorders such as Parkinson’s disease, based on the results of behavioral studies using A2A antagonists (Richardson et al., 1997; Sebastiao and Ribeiro, 1996; Kobayashi and Millhorn, 1999). However, it is still largely unknown how A2A-R modulates cellular functions. Recent studies have shown that activation of A2A-R facilitates calcium-dependent secretion of various neurotransmitters such as GABA, serotonin and glutamate in hippocampal and laterodorsal tegmental cells (Sebastiao and Ribeiro, 2000; Cunha and Ribeiro, 2000a; Mori and Shindou, 2003; Okada et al., 2001; Marchi et al., 2002; Cunha and Ribeiro, 2000b). These results tempted us to dissect the downstream signaling pathways of A2A-R in regulation of calcium-dependent secretion.
The rat pheochromocytoma cell line PC12 cells display phenotypic traits associated with both adrenal chromaffin cells and sympathetic neurons, and is a useful model for studying the calcium-dependent secretion (exocytosis) of neurotransmitters and neuropeptides. In PC12 cells, A2A-R is abundantly expressed, and activation of A2A-R leads to increases in cAMP, as well as activation of PKA, ERK and novel PKC (Chern et al., 1995; Lai et al., 1997; Seidel et al., 1999; Arslan and Fredholm, 2000; Schulte and Fredholm, 2003). It has been shown that NGF enhances neurotransmitter release in PC12 cells through activation of ERK and phosphatidylinositol-3 kinase (PI3K) (Amino et al., 2002). However, it remained unclear whether activation of A2A-R facilitates calcium-dependent secretion in PC12 cells, and if so, which signaling pathway is responsible for this phenomenon downstream of this receptor.
In this study, we monitored the calcium-evoked secretion in PC12 cells by the use of a fusion protein between neuropeptide Y and a modified yellow fluorescence protein (NPY-Venus) (Nagai et al., 2002) and found that activation of A2A-R by its selective agonist facilitates secretion of NPY-Venus upon depolarization. We further dissected the downstream pathways and found the involvement of the PI3K and PKA pathways, possibly through the recruitment of secreted vesicles to the vicinity of the plasma membrane.
The NPY-Venus-expressing plasmid was a kind gift of Dr. A. Miyawaki (RIKEN, Japan). A DNA fragment corresponding to the human growth hormone (hGH) gene was isolated from pXGH5 vector (Nichols Institute) and subcloned into BamHI and EcoRI site of pcDNA3 (Invitrogen, Carlsbad, CA, USA). A dominant-negative Akt (3A Akt) was generated by mutating Lys179, Thr308, Ser473 into Ala and subcloned into Bam HI site of pCS2+ (Masuyama et al., 2001). The plasmid expressing protein kinase inhibitor (PKI) was a kind gift of Dr. M. Greenberg (Harvard Medical School, Boston, MA, USA). Antibodies to phospho MAPK (Promega, Madison, WI, USA), to phospho JNK (Cell Signaling Technology, Beverly, MA, USA), to phospho c-Jun (Cell Signaling), to phospho p38 (Cell Signaling), to phospho ATF-2 (Cell Signaling), to phospho Akt (Cell Signaling), to phospho CREB (Cell Signaling), to Akt (Cell Signaling), to CREB (Cell Signaling), to GAPDH (Chemicon, Temecula, CA, USA), and to human growth hormone (Dako, Carpinteria, CA, USA) were used for immunoblot analysis. CGS21680 was obtained from Sigma-Aldrich (St. Louis, MO, USA); KT5720, LY294002, U0126, SB202190 were from Calbiochem (San Diego, CA, USA); SP600125 was from BioMol (Plymouth Meeting, PA, USA).
Cells were washed with phosphate-buffered saline (PBS) and then lysed in an extraction buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM β-glycerophosphate, 5 mM EGTA, 1 mM sodium pyrophosphate, 5 mM NaF, 1 mM Na3VO4, 0.5% Triton X-100, 1 mM dithiothreitol] supplemented with protease inhibitors [1 mM phenylmethylsulfonyl fluoride, leupeptin (5 μg/ml), pepstatin A (5 μg/ml), aprotinin (5 μg/ml)]. The cell lysates were subjected to immunoblot analysis with specific antibodies and the immune complexes were detected with chemiluminescence reagent (PerkinElmer, Boston, MA, USA).
PC12 cells (5×105 cells, the day before transfection) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 5% horse serum at 37°C in 5% CO2. The expression vector encoding the NPY-Venus fusion protein was transfected into PC12 cells with LipofectAMINE Plus reagent (Invitrogen) according to the manufacturer’s instructions. Seventy-two hours following transfection, the cells were trypsinized and plated on 10-cm dishes. The transfected cells expressing the neomycin-resistant gene were selected by using G418 (Nacalai, Kyoto, Japan) at a concentration of 750 μg/ml. Production of NPY-Venus fusion protein in each established cell line was verified by fluorescence.
The PC12 cell clone stably expressing NPY-Venus fusion protein was plated at a density of 3×105 cells per 6 well dish and incubated for 48 h. The culture medium was removed and replaced with DMEM (without phenol red) supplemented with 10% fetal bovine serum and 5% horse serum. After pretreatment of the cells under various conditions as indicated in the figure legends, the cells were incubated for 3 min with a high-KCl solution (85 mM NaCl, 60 mM KCl, 1.2 mM KH2PO4, 2.5 mM CaCl2, 1.2 mM MgSO4, 20 mM HEPES, pH 7.4, and 11 mM glucose) to induce depolarization. The amount of NPY-Venus released into the medium was measured by detecting fluorescence with a SPECTRAmax Gemini (Molecular Devices). This procedure is schematically shown in Fig. 1A.
 View Details | Fig. 1. The A2A-R agonist CGS21680 facilitates depolarization-induced secretion of NPY-Venus in PC12 cells. (A) Scheme of the procedure to examine the effects of CGS21680 and various kinase inhibitors on depolarization-evoked release of NPY-Venus from PC12 cells. (B) NPY-Venus-expressing cells were incubated for 15 min in the presence or absence of 1 μM of CGS21680. The culture medium was then replaced with 4.7 mM KCl-containing salt solution (low KCl), 60 mM KCl-containing salt solution (high KCl) or 0.5% TritonX-100-containing salt solution for 3 min. The resulting medium was collected for detection of NPY-Venus by fluorescence intensity (upper panel), or released LDH by the use of LDH cytotoxicity detection kit (lower panel). Results are presented as mean±SD from triplicate samples. (C and D) PC12 cells stably expressing NPY-Venus were incubated for 15 min in the presence of various concentrations of CGS21680 (C) or for the indicated times in the presence of 1 μM of CGS21680 (D). The culture medium was then replaced with 60 mM KCl-containing physiological salt solution and incubated for 3 min. The resulting medium was collected for detection of secreted NPY-Venus by fluorescence intensity. Results are presented as mean±SD from triplicate samples. Essentially the same results were obtained in three independent experiments. |
The LDH activity in the culture medium was determined as NADH oxidation reduction using a LDH cytotoxicity detection kit (TaKaRa Biomedicals, Tokyo, Japan). Reaction products were then assayed using a spectrophotometer at A490.
PC12 cells were plated at a density of 7.5×105 cells per 6 well dish and incubated for 24 h. The cells were co-transfected with 3 μg of pcDNA3-hGH and 0.5 μg of other expression plasmids using Lipofectamine2000 reagent according to the manufacturer’s instructions (Invitrogen) and incubated for 72 h. The cells were then washed with a physiological salt solution (140 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 2.5 mM CaCl2, 1.2 mM MgSO4, 20 mM HEPES, pH 7.4, and 11 mM glucose) and incubated for 3 min with a high-KCl solution. The amount of hGH released into the medium was measured by immunoblotting. Immunoreactive bands were captured by ATTO Cool Saver (Atto) and analyzed by ATTO CS analyzer software (version 1.0) (Atto).
To analyze the localization of NPY-Venus-containing vesicles, PC12 cells stably expressing NPY-Venus were plated onto poly-D-lysine-coated glass bottom dishes and cultured for 48 h. After stimulation with 1 μM CGS21680, fluorescence images were recorded by confocal laser scanning microscope (LSM-510, Zeiss). For quantitative analysis of the NPY-Venus-containing vesicle localization, the intensities of fluorescence of NPY-Venus were scored by using LSM5 Image Browser. Detailed procedures are described in the legend for Fig. 4C.
Calcium-dependent exocytosis of synaptic vesicles and that of dense-core vesicles are regulated by overlapping but distinct mechanisms. Although activation of A2A-R in general facilitates calcium-dependent exocytosis of synaptic vesicles (with a few exceptions) (Sebastiao and Ribeiro, 2000; Cunha and Ribeiro, 2000a; Mori and Shindou, 2003; Okada et al., 2001; Marchi et al., 2002; Kirk and Richardson, 1994; Kurokawa et al., 1994; Cunha and Ribeiro, 2000b), it has not been investigated so far whether A2A-R activation also facilitates exocytosis of dense-core secretary vesicles. We established a stable cell line of PC12 cells expressing NPY-Venus, a fusion protein between NPY and a yellow fluorescence protein variant relatively resistant to low pH (Venus), which has been shown to be efficiently targeted to dense-core vesicles (Nagai et al., 2002). We first confirmed that depolarization induced by the addition of 60 mM KCl for 3 min resulted in secretion of NPY-Venus into the culture medium, whereas the addition of 5 mM KCl for 3 min did not, and that this depolarization-induced release of NPY-Venus was dependent on extracellular calcium (Fig. 1A, B and data not shown). Importantly, pretreatment of these PC12 cells with the A2A-R agonist CGS21680 enhanced the depolarization-evoked release of NPY-Venus in a dose-dependent manner (Fig. 1B and C). The evoked release of NPY-Venus reached maximal level at around 15 min of CGS21680 pretreatment (~4-fold increase over the control value without CGS21680 pretreatment), and gradually declined over time (Fig. 1D). We confirmed that the addition of neither 60 mM KCl for 3 min nor 1 μM CGS21680 for 15 min resulted in cell lysis as judged by the absence of lactate dehydrogenase (LDH) release into the medium (Fig. 1B). These results suggest that activation of A2A-R facilitates calcium-dependent exocytosis of dense-core vesicles in PC12 cells.
We next asked which signaling molecules are activated downstream of A2A-R in PC12 cells. A2A-R is known to couple to Gsα of trimeric G proteins that activates adenylate cyclase, thereby elevating the level of cyclic AMP and activating PKA in response to ligand binding (Klinger et al., 2002). It has also been reported that activation of A2A-R increases the activity of ERK MAP kinase and PKCζ (Chern et al., 1995; Lai et al., 1997; Seidel et al., 1999; Arslan and Fredholm, 2000; Schulte and Fredholm, 2003). We found that CGS21680 treatment resulted in phosphorylation of p38 and JNK MAP kinases, in addition to that of ERK (Fig. 2). Phosphorylation of Akt was also observed in response to CGS21680 treatment (Fig. 2). Phosphorylation of these kinases reached maximal level in 10–15 min of CGS21680 treatment and declined gradually after that. We then investigated the phosphorylation level of the transcription factors CREB and ATF2, among the substrates for these kinases. Phosphorylation of CREB at Ser133 (which can be catalyzed by kinases such as PKA, Akt, and Rsk) and that of ATF2 at Thr71 (catalyzed by p38 and JNK) were all increased transiently in response to CGS21680 treatment peaking at around 10–15 min (Fig. 2).
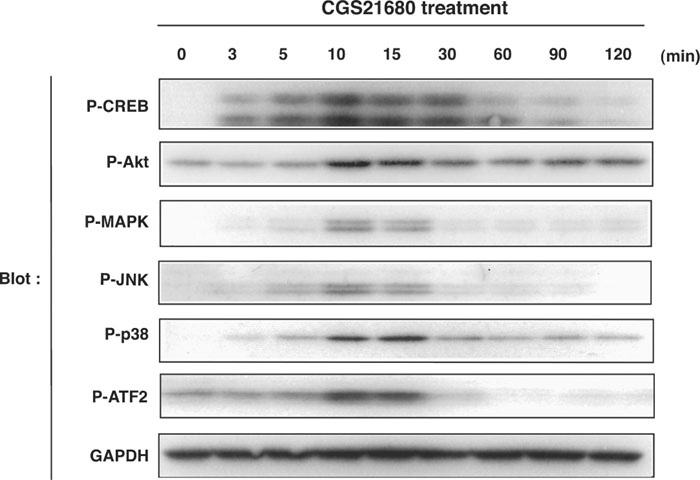 View Details | Fig. 2. Treatment with CGS21680 induces phosphorylation of various signaling molecules in PC12 cells. PC12 cells were cultured for the indicated times in the presence of 1 μM CGS21680, and then lysed and subjected to immunoblot analysis with antibodies to phosphorylated CREB at Ser133, to phosphorylated Akt at Ser473, to phosphorylated ERK at Thr202 and Tyr204, to phosphorylated JNK at Thr183 and Tyr185, to phosphorylated p38 at Thr180 and Tyr182, or to GAPDH as a loading control. |
In an attempt to determine which kinases are responsible for the A2A-R enhancement of calcium-dependent exocytosis, we examined the effects of a number of pharmacological inhibitors in this system. The PC12 cells were pretreated with these inhibitors 30 min before the addition of CGS21680, and the depolarization-induced release of NPY-Venus was measured 15 min after the CGS21680 treatment (see Fig. 1A). Treatment with either the MEK inhibitor U0126 (10 μM), the JNK inhibitor SP600125 (20 μM), the p38 inhibitor SB202190 (10 μM), the PKA inhibitor KT5720 (1 μM), or the PI3K inhibitor LY294002 (20 μM) alone did not markedly affect the CGS21680 facilitation of calcium-dependent release of NPY-Venus. However, treatment with KT5720 (1 μM) and LY294002 (20 μM) together significantly suppressed the effect of CGS21680 by 31.8%±5.6 (p<0.005) (Fig. 3A). The concentrations of LY249002 required for the suppression of the effect of CGS21680 in the presence of KT5720 correlated well with those required for the inhibition of Akt phosphorylation, a downstream effector of PI3K (Fig. 3B and data not shown). Likewise, the concentrations of KT5720 required for the suppression of the effect of CGS21680 in the presence of LY249002 roughly correlated with the suppression of CREB phosphorylation, a downstream effector of PKA and of Akt (Fig. 3B and data not shown). To determine whether PKA and Akt mediate the enhancing effect of CGS21680 on calcium-dependent exocytosis, we introduced protein kinase inhibitor (PKI, a physiological inhibitor of PKA) and a dominant-negative form of Akt (DN-Akt) into PC12 cells together with a growth hormone (GH)-expressing vector. GH is also known to target dense-core vesicles in PC12 cells and is used to monitor calcium-dependent exocytosis (Wick et al., 1993; Oishi et al., 1998). Coexpression of PKI and DN-Akt together, but not expression of PKI or DN-Akt alone, significantly inhibited CGS21680-enhanced, depolarization-induced GH release by 31.4%±9.7 (p<0.025) (Fig. 3C). Collectively, these results suggest that A2A-R activation of the PKA and PI3K-Akt pathways cooperatively facilitates calcium-dependent exocytosis of dense-core vesicles in PC12 cells.
 View Details | Fig. 3. Inhibition of the the PKA and PI3K-Akt pathways suppresses CGS21680 enhancement of depolarization-induced release of NPY-Venus in PC12 cells. (A) PC12 cells stably expressing NPY-Venus were preincubated in the presence (+) or absence of 10 μM U0126, 20 μM SP600125, 10 μM SB202190, 1 μM KT5720, 20 μM LY294002 or DMSO for 30 min, followed by a further 15 min incubation in the presence or absence of 1 μM CGS21680. The culture medium was then replaced with 60 mM KCl containing-physiological salt solution and incubated for 3 min. The resulting medium was collected for detection of secreted NPY-Venus by fluorescence intensity. Results are presented as mean±SD from triplicate samples. Essentially the same results were obtained in three independent experiments. (B) PC12 cells expressing NPY-Venus were preincubated with the indicated inhibitors or DMSO for 30 min, followed by a 15 min incubation with 1 μM CGS21680, and subjected to immunoblot analysis with antibodies to phosphorylated Akt, to Akt, phosphorylated CREB, or to CREB. (C) PC12 cells were transfected with a human growth hormone (hGH)-expressing plasmid together with plasmids encoding protein kinase inhibitor (PKI) or dominant-negative Akt (DN-Akt) for 72 h, followed by a further 15 min incubation with or without 1 μM CGS21680. The culture medium was then replaced with 60 mM KCl containing-physiological salt solution and incubated for 3 min. The resulting medium was collected for detection of secreted hGH by immunoblot analysis. Essentially the same results were obtained in three independent experiments. |
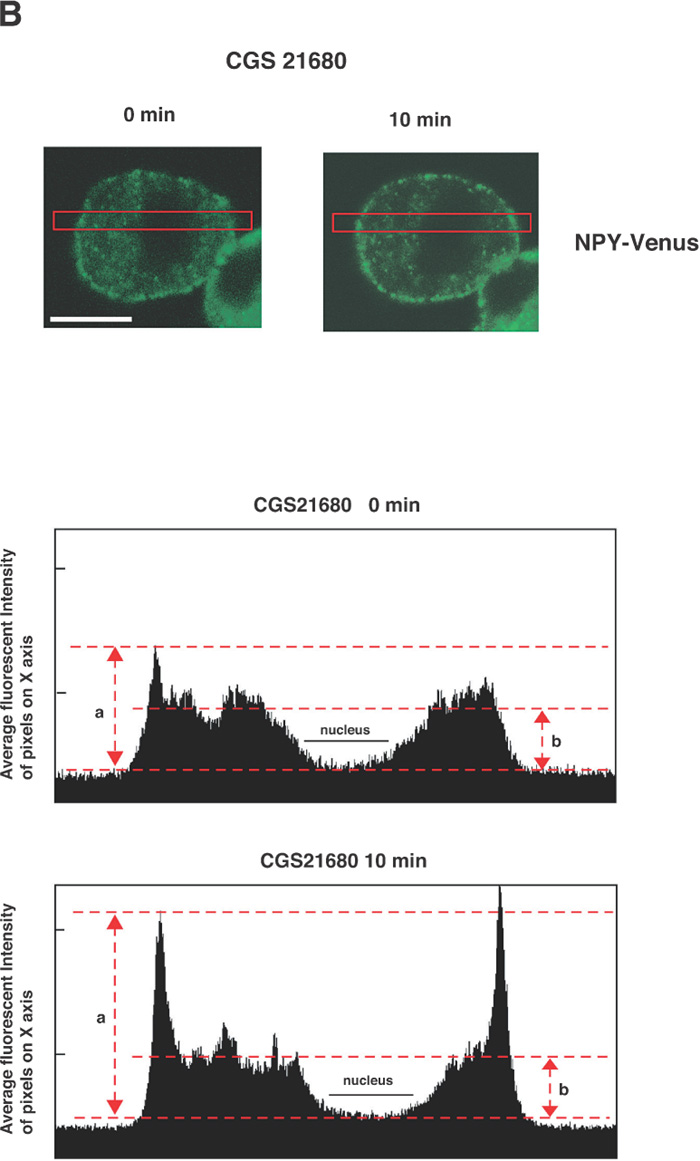
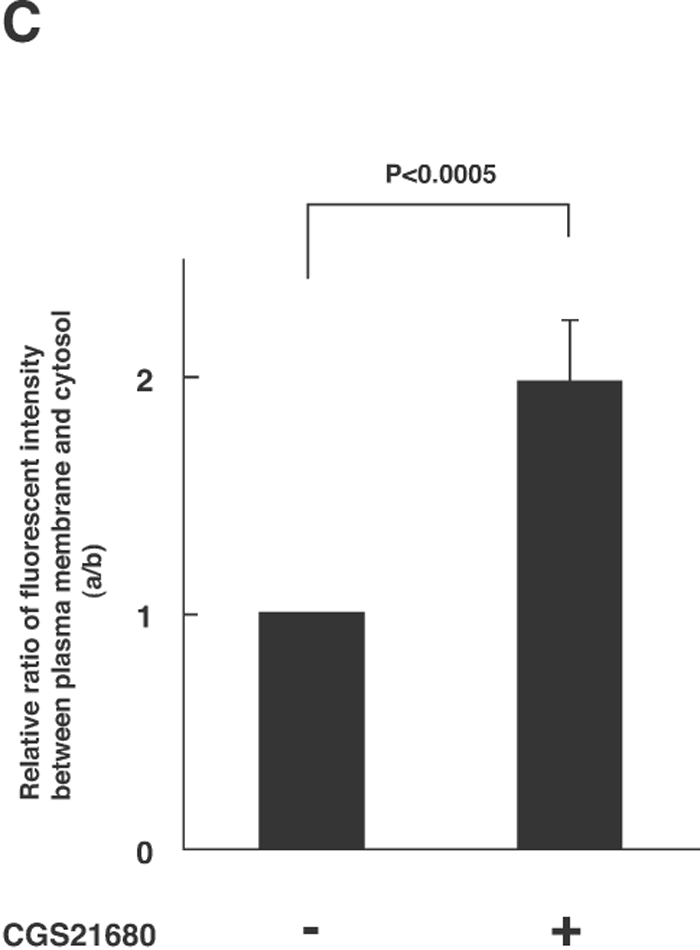
The next key question is how the activation of A2A-R (and its downstream kinases PKA and Akt) enhances the calcium-dependent secretion of NPY-Venus in these cells. One possibility that accounts for this enhancement is to affect the localization of dense-core vesicles and increase the size of the readily releasable pool. We therefore investigated whether the localization of dense-core vesicles is regulated by the treatment with CGS21680, by time-lapse observation of NPY-Venus-containing vesicles. In the absence of CGS21680, the NPY-Venus-containing vesicles were localized dispersedly throughout the cytoplasm (0 min, Fig. 4A, B). However, 10 min after the treatment with CGS21680, a certain proportion of NPY-containing vesicles moved toward the proximity of the plasma membrane, and after 16 min of CGS21680 administration, they appeared aligned beneath the plasma membrane (Fig. 4A, B and online supplemental videos: http://csf.jstage.jst.go.jp). We further carried out quantitative and statistic analysis of these digital images. The ratio of the average fluorescence intensity near the plasma membrane to that in the cytoplasmic regions was calculated from 12 independent images for both CGS21680-treated and -untreated PC12 cells. As shown in Fig. 4C, this ratio was significantly higher in CGS21680-treated cells compared to that in untreated cells (p<0.0005, Student’s t-test). These results clearly indicate that NPY-Venus-containing vesicles translocate to the vicinity of plasma membrane by the exposure of PC12 cells to CGS21680. Interestingly, the alignment of NPY-containing vesicles near the plasma membrane gradually disappeared after 30 min of CGS21680 treatment (Fig. 4A). Thus the time course of the CGS21680-induced recruitment of NPY-Venus-containing vesicles to the proximity of the plasma membrane was closely related to the activation time course of downstream kinases of A2A-R. These results are consistent with the notion that this recruitment of dense-core vesicles may contribute to the enhanced secretion by CGS21680 treatment.
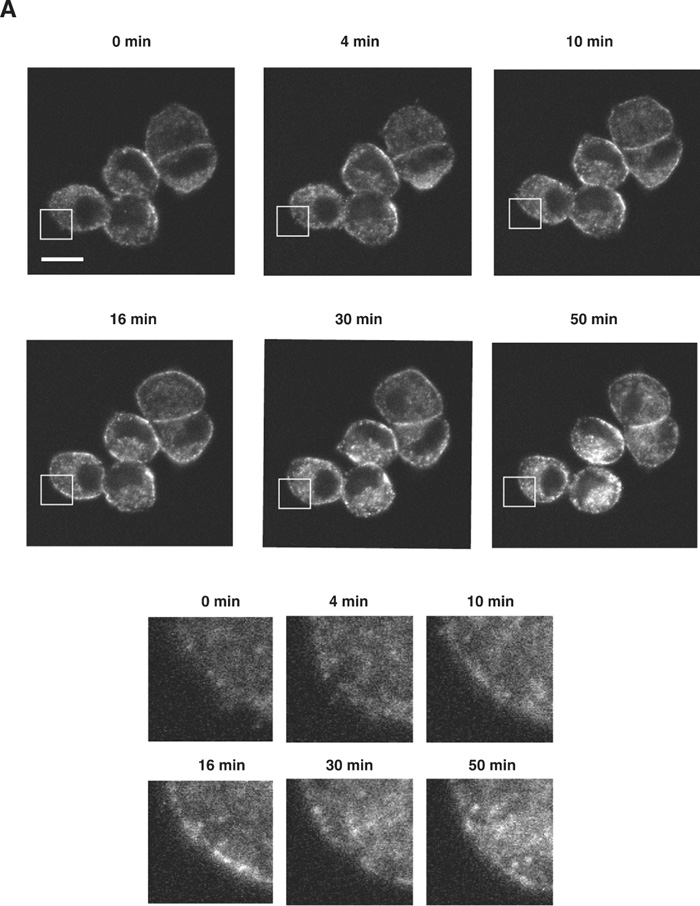 View Details | Fig. 4A. CGS21680-induced translocation of NPY-Venus-containing vesicles to the periphery of PC12 cells. PC12 cells stably expressing NPY-Venus were plated on poly-ABCDECGH-lysine-coated, glass-bottom dishes and cultured for 48 h. The time-lapse fluorescence images were obtained by the use of confocal laser scanning microcrope (LSM510, Zeiss) and analyzed by LSM5 Image Browser. (A) Typical time-lapse images of cells after stimulation with 1 μM CGS21680 are shown. Note that NPY-Venus-containing vesicles were aligned beneath the plasma membrane ~10 min after CGS21680 stimulation. Online supplemental videos are available at http://csf.jstage.jst.go.jp; Click “supplementary materials” under abstract to view the movie. Scale bar, 10 μm. (B) The fluorescence intensity was measured by IPLab software. A rectangular region through the middle of the cells along the longitudinal axis was selected (red box, upper panels). The shorter side consists of 50 pixels of the image, which was captured in the minimum confocal aperture. The average intensity of 50 pixels at any point along the x-axis was calculated, and the values were plotted against the x-distance (lower panels). The position of the nucleus is also indicated. Maximum fluorescence intensity for NPY-Venus at the proximity of the plasma membrane significantly increased after stimulation with CGS21680. (C) Quantitative analysis of the intracellular distribution of NPY-Venus. The laser-scanning confocal microscopy images spanning the center of the cell were subjected to image analysis by LSM5 Image Browser. The averaged fluorescence intensity in the nucleus is regarded as the background, and fluorescence intensities of plasma membranes (a) and cytoplasm (b) are defined as the peak fluorescence intensity at the plasma membrane, and the averaged fluorescence intensity of three-fourths distant from the edge of nucleus to the plasma membrane, respectively, as indicated by red broken bars with arrows in (B). The ratio of (a) to (b) was calculated from 12 independent images for both CGS21680-treated and -untreated PC12 cells. Data are means±SD. P<0.0005, Student’s t test. |
 View Details | Fig. 4B. |
 View Details | Fig. 4C. |
In summary, by the use of a selective agonist for A2A-R, we found in this study that (1) A2A-R facilitates calcium-dependent exocytosis of dense-core vesicles, (2) A2A-R induces phosphorylation of Akt, p38, JNK, CREB and ATF2 (in addition to the previously known downstream targets PKA and ERK), (3) A2A-R enhancement of calcium-dependent secretion is in part mediated by activation of PKA and PI3K-Akt, as revealed by specific pharmacological and physiological inhibitors, (4) A2A-R promotes recruitment of NPY-Venus-containing vesicles to the proximity of the plasma membrane, which may contribute to the facilitation of secretion. Since the PI3K and PKA inhibitors had little effect on the CGS21680-induced translocation of NPY-Venus-containing vesicles in our preliminary experiments, A2A-R might regulate this event independently of the PI3K and PKA pathways.
Akt and PKA share common substrates as they have similar phosphorylation consensus motifs. The cooperative effects between Akt and PKA in mediating the effect of A2A-R activation might be due to phosphorylation of common targets in the process of calcium-dependent secretion of dense-core vesicles.
This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan, and by PRESTO21 and CREST of the Japan Science and Technology Corporation.
|