| To whom correspondence should be addressed: Yoshihiro Yoneda, Laboratories for Biomolecular Networks, Department of Frontier Biosciences, Graduate School of Frontier Biosciences, Osaka University, 1-3 Yamadaoka, Suita, Osaka 565-0871, Japan. TEL: +81–6–6879–4605, FAX: +81–6–6879–4609 E-mail: yyoneda@anat3.med.osaka-u.ac.jp Abbreviations: RanGAP1, Ran GTPase activating protein 1: RCC1, regulator of chromosome condensation 1; RanBP1, Ran binding protein 1; RanBP2, Ran binding protein 2; MALDI-TOF-MS, matrix assisted laser desorption/ionization time of flight mass spectrometry; Ubc9, ubiquitin conjugate enzyme 9; FBS, fetal bovine serum; HEPES, N-(2-hydroxyethyl) piperazine-N'-(2-ethanesulfonic acid); GST, glutathione-S-transferase; FPLC, fast protein liquid chromatography; PMSF, phenylmethane sulfonyl fluoride; PAGE, polyacrylamide gel electrophoresis. |
Ran is a Ras-related small GTPase that is required for nuclear transport, RNA processing, cell cycle control, mitotic spindle formation, and postmitotic nuclear assembly (Bischoff et al., 1995a; Kalab et al., 1999; Nishimoto, 1999). Like other Ras-related GTPases, Ran exists as GTP- and GDP-bound forms. Essential regulators that catalyze Ran to cycle between these two different forms are a Ran-specific GTPase-activating protein RanGAP1 (Bischoff et al., 1994) and a chromatin-bound nucleotide exchange factor RCC1 (Nemergut et al., 2001). In addition to these factors, a cytoplasmic protein RanBP1 and a nucleoporin RanBP2/Nup358 further stimulate the GTPase activity of Ran through the direct binding to Ran-GTP (Bischoff et al., 1995b). Stimulation by RanBP1 and RanBP2/Nup358 is thought to be required for the release of Ran from the blockage of the GTPase activity, which is mediated by the binding of importin-β type transport receptors to Ran-GTP during nucleocytoplasmic transport (Bischoff and Gorlich, 1997).
RanGAP1 contains an evolutionarily conserved domain organization, i.e., N-terminal 11 leucine-rich repeats (LRRs) followed by a stretch of acidic residues and the COOH-domain (Hilling et al., 1999). Structural analysis of co-crystals of yeast homologues of Ran (Gsp1p) and RanGAP1 (Rna1p) revealed that charged residues within the LRR domain of RanGAP1 and Ran mediate interactions between Ran and RanGAP1 (Seewald et al., 2002). By binding to Ran-GTP, RanGAP1 increases the intrinsic GTPase activity of Ran more than 105-fold (Klebe et al., 1995). The functional roles of the acidic domain of RanGAP1 continue to remain controversial, despite the fact that the domain is conserved over different phyla. The acidic domain of yeast rna1p was reported to be essential for Ran-GTP binding and/or RanGAP1 activity (Haberland et al., 1997, Traglia et al., 1989). A recent report, however, indicated that the acidic region of yeast Rna1p was dispensable for GAP activity and Ran binding, but instead, was essential for microtubule formation during mitosis (Seewald et al., 2003).
Vertebrate RanGAP1 is localized in the cytoplasm during interphase. Most of them stably associate with the cytoplasmic face of the nuclear pore complex (NPC) via an interaction with RanBP2/Nup358. The interaction requires a post-translational modification of RanGAP1 with a small ubiquitin-related modifier SUMO1 at the COOH-domain. In plant cells, a RanGAP1 homologue is localized at the NPC through an alternative interaction between the NPC and the N-terminal WPP motif, which is comprised of a trp-pro-pro tripeptide and is conserved among plant RanGAP proteins, albeit no plant homologue of RanBP2 has been identified (Jeong et al., 2005). Thus, the interaction between RanGAP1 and the NPC is evolutionarily conserved. In interphase cells, the localization of RanGAP1 and RCC1, which is compartmentalized in the nucleus as a result of a stable association with chromatin, results in the formation of a Ran-GTP gradient between the nucleus and the cytoplasm, where most of the cytoplasmic Ran is in the GDP-bound state. During mitosis, the interaction was reported to be essential for interaction with microtubules and kinetochores (Joseph et al., 2004). The fact that the SUMO1 modification of RanGAP1 is reversible in cell extracts raises the possibilities that the localization of RanGAP1 is a dynamic process and that it may be regulated, by some currently unknown mechanism. Indeed, Swaminathan et al. recently reported that SUMO1 modified RanGAP1 is phosphorylated at amino acid residues 409T, 428S and 442S during mitosis. The phosphorylated RanGAP1 remains associated with RanBP2/Nup358 and the SUMO E2 conjugating enzyme Ubc9 during mitosis, hence functional relationships between the phosphorylation and RanGTPase cycle and/or RanBP2-dependent SUMO1 modification can be inferred (Swaminathan et al., 2004).
We have observed that the RanGAP1 protein is phosphorylated by cytoplasmic extracts prepared from asynchronous cells. In this report, we demonstrate that the 358S residue of RanGAP1, which is localized within the acidic domain, is phosphorylated. In addition, we found that the 358S phosphorylated RanGAP1 was able to form a stable ternary complex with Ran and RanBP1 in vivo, suggesting that the nucleocytoplasmic transport may be regulated through the phosphorylation of RanGAP1.
The plasmid pRSET-RanGAP1 was constructed by inserting a cDNA encoding human RanGAP1 (GenBank accession NM_ 002883) that had been amplified by RT-PCR using oligonucleotide primers 5'-GTTCCGGGATCCATGGCCTCGGAAGACATTGCCAAG-3' and 5'-GTTGCAGAATTCCTAGACCTTGTACAGCGTCTGCAG-3' into the BamHI/EcoRI site of the pRSET-A vector (Invitrogen, Carlsbad, CA, USA). Plasmid pGEX-6p-RanGAP1 was constructed by inserting a 1.8 kbp BamHI/EcoRI fragment of the RanGAP1 cDNA isolated from pRSET-RanGAP1 into pGEX-6P1 (Amersham Bioscience, Piscataway, NJ, USA). Plasmid pFLAG-CMV2 RanGAP1 was constructed by inserting a 1.8 kbp BamHI/SmaI fragment from pGEX-6p-RanGAP1 into the pFLAG-CMV2 vector (Sigma-Aldrich, St. Louis, MO, USA). pCAG-wtag-RanGAP1 was constructed by inserting a 1.8 kbp BamHI/XhoI fragment of RanGAP1 cDNA from pGEX-6p-RanGAP1 into pCAG-wtag. The construction of the pCAG-wtag vector will be described elsewhere. The RanGAP1S356AS358A, RanGAP1S356A, RanGAP1S358A, RanGAP1S358T, RanGAP1R91A and RanGAP1K524R were constructed using a Quick-change site directed mutagenesis kit (Stratagene, La Jolla, CA, USA) using plasmid pRSET-RanGAP1 as template. The following sense and anti-sense oligonucleotide pairs were used to introduce each mutation; S358A: (5'-GGTGCTGGCGagtCTCgcTGATGACGAGGACGAGGAGG-3' and 5'-CCTCCTCGTCCTCGTCATCAgcGAGactCGCCAGCACC-3'), S358T: (5'-GGTGCTGGCGagtCTCAcTGATGACGAGGACGAGGAGG-3' and 5'-CCTCCTCGTCCTCGTCATCAgTGAGactCGCCAGCACC-3'), R91A: (5'-GCCACTGGAGTGAtATGTTCACGGGAgcGCTGCGGACCG-3' and 5'-CGGTCCGCAGCgcTCCCGTGAACATaTCACTCCAGTGGC-3'), K524R: (5'-GGGTCTGCTCAgGAGtGATGACAAGGTCAAGGCC-3' and 5'-GGCCTTGACCTTGTCATCaCTCcTGAGCAGACCC-3'), S356AS358A: (5'-GGCCAAGGTGCTGGCGgCCCTCgcTGATGACGAGGACG-3' and 5'-CGTCCTCGTCATCAgcGAGGGcCGCCAGCACCTTGGCC-3'), S356A: (5'-GGCCAAGGTGCTGGCGgCCCTCAGTGATGACGAGGACG-3' and 5'-CGTCCTCGTCATCACTGAGGGcCGCCAGCACCTTGGCC-3'). The sequences were verified by DNA sequencing.
The GST-SUMO1 protein expressed from pGEX-SUMO-1GG vector (G97) has a phosphorylation site derived from pGEX-2TK (Saitoh et al., 1998). Thus, the cDNA encoding SUMO-1 GG (G97) was amplified by PCR using the following oligonucleotides: 5'-GCGGATCCTCTGACCAGGAGGCCAAACCTTCAACTG-3', (a BamHI restriction site is underlined) and 5'-CGCCCGGGTTAACCCCCCGTTTGTTCCTGATAAACTTCAATCAC-3', (a SmaI restriction site is underlined) and subcloned to pGEX-6P3 (Amersham Pharmacia) to remove the phosphorylation site.
All the oligonucleotides indicated above were synthesized by the Japan Bio Services Co., Ltd. (Saitama, Japan).
HeLa and HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C in 5% CO2 atmosphere. Transfection into HeLa and HEK293T cells was performed by means of an Effectene Transfection Reagent Kit according to the manufacturer’s recommended protocol (Qiagen, Hilden, Germany). Ehrlich cytosolic extract was prepared as described previously (Hieda et al., 2001).
HeLa cells expressing GST- or FLAG-RanGAP1, their mutants, and pEF-GST-STAT3711–770 (Abe et al., 2001) were pre-incubated for 3 hr in DMEM supplemented with 10% FBS dialyzed against HEPES buffered saline (25 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM KCl, 1 mM dithioerythritol). After washing with HEPES buffer, the cells were further incubated in DMEM supplemented with 10% dialyzed FBS containing 0.5 mCi/ml of [32P]-phosphorus (Amersham Biosciences) for 3 to 24 hr.
Cells expressing FLAG-RanGAP1, GST-RanGAP1 and their mutants were washed with HEPES buffer and lysed by a lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Triton X-100, 1 mM dithioerythritol, 0.4 mM PMSF, 5 μg/ml each of aprotinin, leupeptin, and pepstatin A; 5 mM NaF, 1 mM Na3VO4). After homogenization and centrifugation at 20,000×g for 15 min at 4°C, the total soluble lysates were mixed with anti-FLAG M2 agarose (Sigma-Aldrich) or glutathione Sepharose 4B beads and rotated for 2 hr at 4°C. The beads were washed with lysis buffer and bound proteins were then eluted by the addition of SDS-PAGE sample buffer (Fig. 1, 2A) or lysis buffer containing 1 mg/ml of 3×FLAG peptide (Sigma-Aldrich) (Fig. 5B, C).
 View Details | Fig. 1. RanGAP1 is phosphorylated in vivo. (A) HeLa cells expressing GST- or FLAG-tagged RanGAP1 proteins were incubated in DMEM-10% FCS containing 0.5 mCi/ml of [32P]-phosphorus for 24 hr. After purification with glutathione Sepharose or anti-FLAG-agarose beads, the fusion proteins were resolved by 10% SDS-PAGE and stained with Coomassie brilliant blue (CBB) (lower panel). Phosphorylation of RanGAP1 was detected by autoradiography (upper panel). Phosphorylation of a fragment of STAT1 protein (lane 4) was served as a positive control. Extracts prepared from cells that had been transfected with an empty vector (–) were used as negative control. (B) FLAG-RanGAP1 proteins purified from HeLa (top panel) and E. coli (lower panel) cells were resolved by 10% SDS-PAGE. Each protein band was excised from the gel, digested by trypsin and the peptide mass determined by MALDI-TOF-MS. Shown are the non-phosphorylated (white arrow) and phosphorylated (black arrow) peptide peaks. The amino acid sequence of the peptide fragment is indicated below the panels. The underlines denote predicted phosphorylation sites. (C) HeLa cells expressing FLAG-tagged wild type (wt) and mutant (S356A-S358A, S356A, S358A-S358T) RanGAP1 proteins were labeled with [32P]-phosphorus for 3 hr as in Figure 1. After purification with anti-FLAG-agarose beads, the FLAG-RanGAP1 proteins were resolved by 10% SDS-PAGE and stained with CBB (bottom panel). The phosphorylation of each protein was analyzed by autoradiography (top panel). A sample prepared from cells transfected with an empty vector was used as a negative control. |
 View Details | Fig. 2. Casein kinase II (CK2) is the specific kinase for 358S of RanGAP1. (A) HeLa cells expressing wild type or S358A FLAG-RanGAP1 proteins were treated with CK2 inhibitors, apigenin or DRB, for 2 hr and further incubated in the presence of [32P]-phosphorus and the CK2 inhibitors for 3 hr. After being purified with anti-FLAG-agarose beads, wild type and S358A FLAG-RanGAP1 proteins were resolved by 10% SDS-PAGE and stained with CBB (bottom panel). Phosphorylation was examined by autoradiography (top panel). The incorporation of radioactivity into each band was determined by means of an imaging analyzer. Degrees of phosphorylation of RanGAP1 under different conditions are indicated relative to that in the absence of CK2 inhibitor, which is arbitrarily set at 1. (B) Bacterially expressed GST-tagged wild type or S358A RanGAP1 proteins were incubated with a HeLa cell cytosolic fraction and [γ-32P]ATP in the presence or absence of apigenin at 25°C for 30 min. After purification with glutathione Sepharose beads, the bound proteins were resolved by 10% SDS-PAGE, stained with CBB (left panel), and subjected to autoradiography (right panel). GST alone was used as a negative control. (C) Recombinant GST-tagged wild type and S358A RanGAP1 proteins were phosphorylated in the presence of [γ-32P]ATP at 25°C for 30 min. In lanes 4, 5, 7, 8, phosphorylation reactions were done in the presence of CK2 inhibitors. After resolving the protein bands by 10% SDS-PAGE, they were detected by CBB staining (left panel) followed by autoradiography (right panel). GST and dephosphorylated casein were used as negative and positive controls, respectively. Incorporation of radioactivity to each band in the gel is indicated as in Figure 2A. |
Plasmids encoding GST-fusion proteins (GST-RanGAP1 and their mutants) were introduced in E. coli BL21(DE3) and the proteins were expressed by adding 1 mM of isopropyl-1-thio-β-D-galactopyranoside to the culture medium. Cells were harvested, extracted by sonication, and the lysates were passed through glutathione Sepharose 4B (Amersham Biosciences) columns according to the manufacturer’s recommended protocol. The GST fusions were cleaved from the columns by treatment with PreScission Protease (Amersham Biosciences) overnight at 4°C. After passing through PD-10 columns (Amersham Biosciences), that had been previously equilibrated with buffer A (20 mM HEPES (pH 7.3), 150 mM NaCl, 1 mM 2-mercaptoethanol), the wild type and mutant RanGAP1 proteins were applied to a MonoQ FPLC column (Amersham Biosciences) equilibrated with buffer A. A linear gradient of 0.15 to 1 M NaCl in buffer A was applied, and RanGAP1 and the mutant proteins that eluted between the 0.6 and 0.7 M NaCl fractions were pooled, exchanged with HEPES buffer (25 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM KCl, 1 mM dithioerythritol) by means of a PD-10 column.
In some cases, the GST-fusion proteins (GST-SUMO1, GST-RanGAP1 and the mutants) were expressed and bound to glutathione Sepharose 4B columns as above and then eluted with elution buffer (10 mM glutathione (Sigma-Aldrich), 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 1 mM dithioerythritol). Relevant fractions, identified by immunoblot using an anti-GST antibody, were exchanged with protein kinase assay buffer (50 mM Tris-HCl (pH 7.4), 30 mM KCl, 5 mM MgCl2, 1 mM dithioerythritol) by PD-10 column chromatography.
Ran was expressed in E. coli BL21 (DE3) from the pET3d vector (Dasso et al., 1994). RanGTP was purified as described previously (Bischoff et al., 1994). The purified proteins were shock-frozen and stored at –80°C until used. Protein concentrations were determined by a CBB Protein assay kit (Nacalai Tesque, Kyoto, Japan).
To identify the RanGAP1 phosphorylation site, mass analysis was performed by the APRO Life Science Institute, Inc. Both the HeLa cell expressed FLAG-RanGAP1 and bacterially expressed untagged RanGAP1 proteins were resolved by SDS-PAGE, in-gel-digested with trypsin, the peptides analyzed by mass fingerprinting using MALDI-TOF-MS, Voyager-DE STR (Applied Biosystems, Foster City, CA, USA). Peptide mass fragments derived from HeLa cell and E. coli expressed RanGAP1 proteins were compared to determine the phosphorylation site. The predicted peptides were identified using the Mascot Search Engine (www.matrixscience.com).
CK2 inhibitors, apigenin (Sigma-Aldrich) and 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB; Sigma-Aldrich) were dissolved in dimethyl sulphoxide (DMSO), and diluted with assay buffer (50 mM Tris-HCl (pH 7.5), 30 mM KCl, 5 mM MgCl2, 1 mM dithioerythritol, 5 mM NaF, 1 mM Na3VO4) at the time of use. In mock samples, an equivalent amount of DMSO was added to assay buffer. For the phosphorylation reaction, 5 μg of GST, wild type and mutant GST-RanGAP1 proteins were combined with glutathione Sepharose 4B beads, incubated in the presence of 0.1 mM [γ-32P]-ATP (1 μCi) at 25°C for 30 min with or without the cytosolic fraction and 40 μM apigenin or 40 μM DRB. For phosphorylation by CK2, 5 μg of GST, GST-RanGAP1, the mutant RanGAP1 and casein (dephosphorylated, Sigma-Aldrich) were incubated with 25 ng/ml (60 μU) of CK2 (casein kinase 2, active, Upstate USA, Chicago, IL, USA) in assay buffer containing 0.1 mM [γ-32P]-ATP (1 μCi) with or without the inhibitors at 25°C, for 15 min.
Monoclonal antibodies against RanGAP1 (Zymed, San Francisco, CA, USA), Ran, RanBP1 (Transduction Laboratories, San Diego, CA, USA), and FLAG M2 peptide (Sigma-Aldrich) were purchased from the above companies.
Immunization of rabbits with peptides LASLpSDDEDE (pS indicates phospho-serine) and LASLSDDEDE, bleeding, and the affinity purification of antibodies by each peptide column was performed by Sigma Genosys. The resulting antibodies, that recognized the phosphorylated or unphosphorylated forms of RanGAP1, were designated as anti-pS358 and anti-S358, respectively.
For the anti-pS358 and anti-S358 antibodies, nitrocellulose membranes blotted with proteins were treated with Blocking-One blocking reagent (Nakalai Tesque) and incubated with anti-pS358 (2 μg/ml) or anti-S358 antibodies (5 μg/ml) in 0.05% Tween 20/PBS (PBST) containing 20% Blocking-One for 1 hr. The membrane was washed with PBST, and incubated for a further 45 min with the anti-rabbit secondary antibody (Bio-Rad, Hercules, CA, USA) diluted in PBST containing 20% Blocking-One. After washing with PBST, signals were developed by ECLTM Western Blotting Detection Reagents (Amersham Biosciences) followed by exposure to X-ray films.
For the other antibodies, the blotted membranes were blocked in PBST containing 5% skim milk, and incubated for 1 hr with primary antibodies diluted at 1,000–5,000 fold in PBST containing 3% skim milk. The membranes were washed with PBST, and incubated for 45 min with the appropriate secondary antibodies diluted in PBST containing 3% skim milk. After washing with PBST, the signals were detected, as described above.
GAP assays were performed as described previously (Burstein et al., 1991; Richards et al., 1995) with following modifications. Reactions were carried out at 25°C and terminated by vacuum filtrating onto nitrocellulose membranes (Millipore, Billerica, MA, USA). After washing with wash buffer (40 mM Tris-HCl (pH 8.0), 8 mM MgCl2, 2 mM GTP), [γ-32P] the counts remaining on filters were quantitated by a liquid-scintillation counting (Perkin-Elmer, Boston, MA, USA).
The wild type and S358A mutant FLAG-RanGAP1 proteins were expressed in HEK293T cells and purified as described above. The purified proteins were bound to anti-FLAG agarose at 4°C for 1 hr. After washing with lysis buffer, Ehrlich cytosolic extracts with or without GST and GST-SUMOGG97 were added to the reaction solutions. They were incubated at 25°C for 30 min and washed with lysis buffer, and the reactions were terminated by adding SDS-PAGE sample buffer. The reactions were subjected by SDS-PAGE followed by immunoblot using an anti-FLAG antibody.
We noted that recombinant RanGAP1 is efficiently phosphorylated in vitro by cell extracts prepared from cultured asynchronous cells (data not shown; see also Fig. 2). To determine whether RanGAP1 is actually phosphorylated in vivo, HeLa cells expressing GST- or FLAG-tagged RanGAP1 were labeled with [32P]-phosphorus and the fusion proteins were purified by glutathione Sepharose or an anti-FLAG agarose resin. As detected by autoradiography, both the GST- and FLAG-tagged RanGAP1 proteins were phosphorylated under these conditions (Fig. 1A). On the other hand, the fusion proteins were not efficiently modified by SUMO1. These results are not consistent with a previous report showing that about half of the endogenous RanGAP1 was modified by SUMO1 (Bischoff et al., 1995b). It is possible that the amounts of cellular SUMO1 and/or the enzymes involved in SUMO1 modification may be a limiting factor, since an excess amount of RanGAP1 was overexpressed (see also below).
To determine which amino acid residue(s) of RanGAP1 are phosphorylated, peptide mass fingerprinting was performed. A peptide peak, which exhibited m/z 5947 in the E. coli-derived preparation, was shifted to 6027 in the HeLa cell-derived RanGAP1 (Fig. 1B). The molecular mass of the E. coli-derived peptide matched a peptide fragment corresponding to residues 353-402 of RanGAP1. The peak identified in the HeLa cell-derived FLAG-RanGAP1 preparation agreed well with the theoretical molecular mass of the corresponding peptide to which one phosphate group was covalently attached (black arrow).
The peptide fragment (353-402) contained two possible phosphorylation sites (356S and 358S). To determine which serine residue is phosphorylated, expression vectors harboring alanine substitution mutations at 356S (S356A), 358S (S358A) or both (S356AS358A) were constructed and their phosphorylation was examined. The phosphorylation of the S356AS358A and S358A mutants was markedly reduced compared with the wild type RanGAP1 (Fig. 1C, lanes 2, 3 and 5), whereas S356A was phosphorylated as efficiently as the wild type protein (Fig. 1C, lane 4). These data indicate that 358S is the major phosphorylation site in vivo. Since a threonine substitution mutant for serine 358 (S358T) was still phosphorylated efficiently (Fig. 1C, lane 6), the kinase involved in the phosphorylation of 358S is assumed to be a serine/threonine kinase.
The amino acid sequence including 358S of RanGAP1, LSDDED, matched the consensus sequence for casein kinase 2 (CK2) (E/D/x-pS/T-D/E/x-E/D/x-E/D-E/D/x; single letter code, x is any amino acid, pS/T is the phosphorylation site). To confirm this hypothesis experimentally, the phosphorylation was analyzed in the presence of the known CK2 inhibitors, apigenin (Song et al., 2000; Song et al., 2003; Farah et al., 2003) and DRB (Zandomeni et al., 1986; Farah et al., 2003) in the culture medium. For the cultured cell lines, apigenin was used in the range of 40 to 150 μM, at which concentrations approximately 50% of the CK2 activity was reported to be inhibited (Song et al., 2003; Song et al., 2000). As shown in Fig. 2A, both drugs inhibited the RanGAP1 phosphorylation in vivo in a dose-dependent manner. As reported previously, higher concentrations of DRB were required to achieve a similar degree of inhibition (Farah et al., 2003). As exemplified in Fig. 2B, the same site of RanGAP1 was also phosphorylated by the HeLa cell-extract in in vitro conditions. In addition, the phosphorylation was inhibited by 40 μM apigenin and DRB (Fig. 2B). The wild type RanGAP1, but not S358A, was consistently phosphorylated by purified CK2 more efficiently than a control substrate, casein (Fig. 2C, lanes 3 and 6). While the CK2 inhibitors, under our in vitro conditions, did not completely eliminate RanGAP1 phosphorylation, it was reduced (Fig. 2C, lanes 3–5). These results suggest that CK2 phosphorylates 358S in RanGAP1.
The phosphorylation site of RanGAP1 was located at the border between the LRR domain and the acidic region of human RanGAP1 (Fig. 3A). The amino acid sequence of this region is highly conserved among vertebrate RanGAP1 proteins. Although the domain is not highly conserved in yeast RanGAP (Rna1p, S. cerevisiae; rna1p, S. pombe), nematode C. elegans Ran-2 and Drosophila RanGAP1, they harbor a serine or threonine residue within the acidic region. On the other hand, in plant RanGAPs [A. thaliana RanGAP1(414S), RanGAP2(419S), M. sativa RanGAP(419S), and rice RanGAP(435S)] (Jeong et al., 2005), the corresponding serine residue appears to exist between the LRR domain and the acidic region. These data suggest that the phosphorylation of RanGAP1 at the border between the LRR domain and the acidic region may play a role in RanGAP1 function.
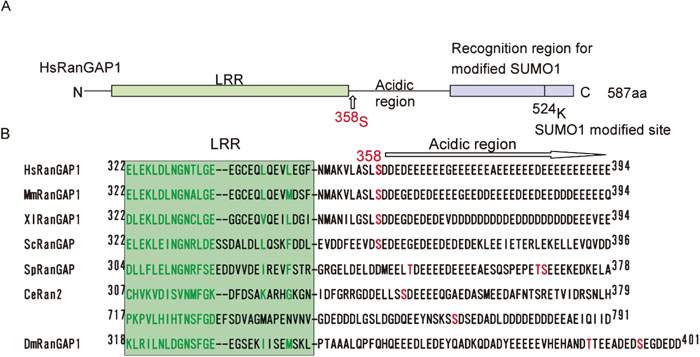 View Details | Fig. 3. 358S of RanGAP1 is conserved from yeast to human. (A) Structural motifs of the human RanGAP1 protein. The domain organization of the human RanGAP1 protein is schematically represented as previously reported (Hilling et al., 1999). (B) Putative phosphorylation sites of RanGAP1 proteins from different species are aligned along with the human RanGAP1 sequence (Hilling et al., 1999, Seewald et al., 2002). CK2 recognition motifs are highlighted (red). The first two letters of the sequence names indicate the organisms as follows: Hs, Homo sapiens; Mm, Mus musculus; Xl, Xenopus laevie; Sc, Saccharomyces cerevisiae; Sp, Schizosaccaromyces pombe; Dm; Drosophila melanogaster. |
To determine if the 358S residue of the endogenous RanGAP1 is actually phosphorylated in vivo, antibodies were raised against synthetic peptides containing the phosphorylated and unphosphorylated 358S residue. As shown in Fig. 4A, the antibodies distinguished the phosphorylated form from HeLa cells from the unphosphorylated form of RanGAP1 from E. coli cells (phosphorylation state was confirmed by mass spectrometry; see Fig. 1B), by immunoblotting (Fig. 4A). In addition, the E. coli RanGAP1 protein, which had been subjected to in vitro phosphorylation by purified CK2 (Fig. 4A, lane 3), and the HeLa cell-derived sample, which had been dephosphorylated by alkaline phosphatase (Fig. 4A, lane 4), were also clearly distinguished, confirming the specificity of the antibodies.
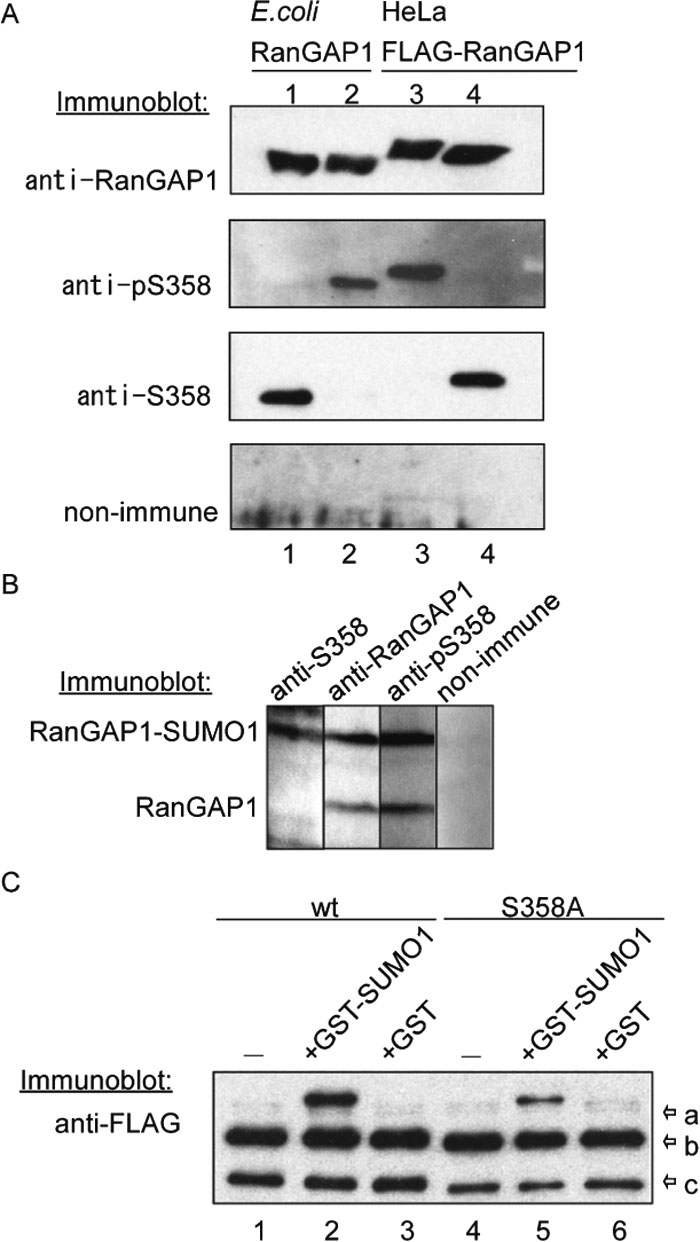 View Details | Fig. 4. 358S of the endogenous RanGAP1 was constitutively phosphorylated and the phosphorylation does not affect efficiency of SUMO1 modification. (A) Bacterially expressed RanGAP1 (lanes 1, 2) and FLAG-RanGAP1 expressed in HeLa cells (lanes 3, 4) were detected by immunoblotting using rabbit antibodies raised against peptides containing phosphorylated (anti-pS358) or non-phosphorylated (anti-S358) 358S residues. Non-immune serum was used as a negative control. In lane 2, bacterially expressed RanGAP1 was phosphorylated in vitro by purified CK2. In lane 4, FLAG-RanGAP1 purified from HeLa cells was dephosphorylated by calf intestine alkaline phosphatase (CIAP). (B) A total extract prepared from HeLa cells was resolved by 10% SDS-PAGE, transferred to nitrocellulose and immunoblotted by anti-S358, anti-RanGAP1 and anti-pS358 antibodies. Non-immune serum was used as a negative control. (C) Wild type and S358A FLAG-RanGAP1 proteins expressed in HEK293T cells were purified with anti-FLAG agarose beads. They were incubated with cytosolic fraction in the absence (–) or presence of GST-SUMO 97GG (GST-SUMO) or GST at 25°C for 30 min and pulled down with anti-FLAG agarose beads. Immunopellets were resolved by 10% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with anti-FLAG antibody. Arrows are shown FLAG-RanGAP1 modified with GST-SUMO (arrow-a), FLAG-RanGAP1 modified with SUMO1 (arrow-b) and FLAG-RanGAP1 (arrow-c). |
The total lysates of asynchronous HeLa cells were analyzed by immunoblot using anti-RanGAP1, anti-pS358 and anti-S358 antibodies (Fig. 4B). Anti-pS358 and anti-S358 antibodies detected endogenous RanGAP1 in cell lysates. Both sumoylated and nonsumoylated endogenous RanGAP1 proteins were phosphorylated at 358S (Fig. 4B, anti-pS358 panel). In contrast, anti-S358 antibody detected sumoylated, but not nonsumoylated, RanGAP1 exclusively (anti-S358 panel), indicating the 358S-dephosphorylated form of RanGAP1 is completely sumoylated. The same results were obtained when cell lysates from HEK293T cells or various mouse cell lines were probed with these antibodies (date not shown).
To examine the possibility that the phosphorylation of RanGAP1 at 358S might affect the efficiency of sumoylation, wild type and S358A FLAG-RanGAP1 proteins expressed in HeLa cells (the wild type FLAG-RanGAP1 protein would be expected to be phosphorylated at 358S) were subjected to an in vitro sumoylation assay (Fig. 4D). Both the wild type and S358A FLAG-RanGAP1 proteins were modified with endogenous SUMO1 and GST-SUMOGG97 to similar extents. Although the phosphorylation site in the RanGAP proteins identified in this study appears to be conserved from yeast to human (Fig. 3B), it is known that only RanGAP from vertebrates is sumoylated in vivo (Hilling et al., 1999). Thus, we conclude that phosphorylation at 358S does not affect the efficiency of sumoylation.
To further examine the functional significance of the phosphorylation of 358S, the GAP activity of wild type RanGAP1 was compared with that of the S358A mutant. The R91A RanGAP1 mutant, which is known to be deficient in Ran-GTP binding (Haberland and Gerke, 1999), was used as a negative control. The activities of the wild type and S358A RanGAP1 proteins were almost the same, regardless of their phosphorylation state (Fig. 5A, upper and lower panels). Since phosphorylated RanGAP1 was prepared under the conditions described in Fig. 4A, it was expected that almost all of the wild type RanGAP1 proteins were phosphorylated. These data, as well as our observation that the intracellular localization of the S358A mutant was similar to that of the wild type protein (data not shown), indicate that phosphorylation at 358S does not significantly alter the known functions of RanGAP1.
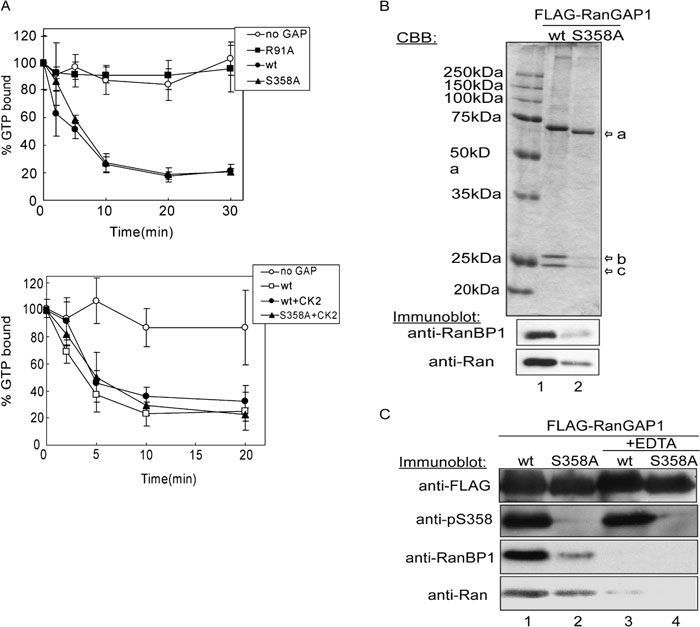 View Details | Fig. 5. Phosphorylation of RanGAP1 at 358S stabilizes a RanGAP1-RanGTP-RanBP1 ternary complex. (A) Upper panel: 1 μM of [γ-32P]GTP-Ran was incubated with 0.75 nM of wild type (closed circle) or S358A (closed triangle) recombinant RanGAP1 proteins at 25°C for 30 min. Recombinant RanGAP1 R91A (closed square), one of the mutants lacking RanGTP binding ability (Haberland and Gerke, 1999), was used for negative control. At each time point, Ran-associated radioactivity was measured by filter binding assay. Data are represented as the mean±S.D. of 4 independent experiments. Lower panel: Same as in A., but recombinant RanGAP1 proteins were in vitro phosphorylated by purified CK2 before GAP assay and incubation was terminated at 20 min. Arrows are shown FLAG-RanGAP1 (arrow-a), RanBP1 (arrow-b) and Ran (arrow-c). (B) Wild type and S358A FLAG-RanGAP1 proteins expressed in HEK293T cells were immunoprecipitated by anti-FLAG agarose beads. Half of each of the eluates were resolved by 10% SDS-PAGE and stained with CBB (top panel). The rest of the samples were transferred to a nitrocellulose membrane after electrophoresis and immunoblotted by anti-RanBP1 (middle panel) or anti-Ran (bottom panel) antibodies. (C) HeLa cells expressing wild type and S358A FLAG-RanGAP1 were lysed in a lysis buffer with or without 10 mM EDTA and immunoprecipitated by anti-FLAG agarose. Proteins were resolved by 10% SDS-PAGE, and transferred to nitrocellulose, and immunoblotted by anti-FLAG (panel 1), anti-pS358 (panel 2), anti-RanBP1 (panel 3) and anti-Ran (panel 4) antibodies. |
Finally, we attempted to determine the nature of the interaction of the phosphorylated form of RanGAP1 with other proteins using a co-immunoprecipitation method. As shown in Fig. 5B, top panel, two RanGAP1-binding proteins with molecular masses ca. 25 kDa were identified. These proteins were not efficiently co-purified with the 358S mutant. To identify these proteins, immunopellets were subjected to immunoblot using a panel of antibodies, which recognize known RanGAP1 binding proteins. As a result, these two proteins were shown to be RanBP1 and Ran (Fig. 5B, lower panels). In addition, as shown in Fig. 5C, the co-immunoprecipitation of Ran and RanBP1 was inhibited by the addition of EDTA to cell extracts, suggesting that complex formation is dependent on the overall structure of Ran, because it is well known that Ran releases the bound nucleotide in the presence of EDTA (Bischoff et al., 1994; Karen and Macara, 1997). These results indicate that the phosphorylated form of RanGAP1 forms a stable complex with RanBP1 and Ran more efficiently than the unphosphorylated form, meaning that the phosphorylation of RanGAP1 apparently affects the stable complex formation with RanBP1 and Ran, suggesting that the Ran cycle may be controlled via the phosphorylation of RanGAP1.
The findings herein demonstrate that RanGAP1 is specifically phosphorylated at 358S. Taking into consideration the facts that two types of known CK2 inhibitors efficiently inhibited the phosphorylation, that purified CK2 actually phosphorylated RanGAP1 in vitro, and that the amino acid sequence, including 358S, matched the consensus sequence for CK2, it is most likely that the kinase responsible for 358S of RanGAP1 is CK2. CK2 is a serine/threonine protein kinase that is distributed ubiquitously in eukaryotic organisms. This kinase is a constitutively active enzyme (Litchfield, 2003) and has been suggested to be essential for various biological processes such as cell cycle progression, cell proliferation, cell survival, signal transduction and pol III related transcription (Litchfield, 2003; Meggio and Pinna, 2003; Hu et al., 2004). However, as far as is known, no reports implicating CK2 in the regulation of nucleocytoplasmic transport have appeared.
We found that most of the endogenous RanGAP1 proteins are constitutively phosphorylated at a serine residue and that the phosphorylation has no impact on the known properties of RanGAP1, including sumoylation (Fig. 4C), GAP activity (Fig. 5A), and intracellular localization (data not shown). It is noteworthy, however, that FLAG-RanGAP1 (probably phosphorylated at 358S), but not the FLAG-S358A mutant, was efficiently co-immunoprecipitated with Ran and RanBP1. A rough calculation from densitometric analysis of the CBB stained gel suggests that the stoichiometry of the complex was about 1:1:1, suggesting that it is a ternary complex. A recent crystallographic study of the yeast RanGAP-Ran-RanBP1 ternary complex indicated that Ran is sandwiched between RanGAP1 and RanBP1, and that RanGAP1 does not directly interact with RanBP1 (Seewald et al., 2003). In addition, it is known that RanBP1 binds to Ran-GTP with several orders of magnitude higher affinity than Ran-GDP, as reported previously (Kuhlmann et al., 1997). Therefore, it is reasonable to speculate that phosphorylation may alter the apparent affinity of RanGAP1 for Ran-GTP thus regulating the Ran cycle. In an in vitro kinetic study using a fluorescence-based test system, Seewald et al. concluded that the binding of RanBP1 to Ran does not significantly affect GTP cleavage or the Pi release steps of the GTPase reaction, but does increase the already very fast Ran-RanGAP association reaction (Seewald et al., 2003). The structural data suggest that the acidic domain of RanGAP interacts with the basic patch in Ran (Seewald et al., 2002). It is therefore conceivable that the addition of phosphate near the acidic region increases the local net negative charge of the domain, thus enhancing its affinity to Ran. Alternatively, phosphorylation induces gross structural changes in the region and stabilizes the interaction. Taking into account the facts that RanGAP1 appears to be phosphorylated during interphase and that SUMO-modified RanGAP1 exists in both the 358S phosphorylated and unphosphorylated forms, it is reasonable to speculate that phosphorylation may alter the apparent affinity of RanGAP1 for Ran-GTP to regulate the Ran cycle, thus affecting nucleocytoplasmic transport. It is known that CK2 is localized in both the nucleus and the cytoplasm (Faust and Montenarh, 2000). Since most of the exogenously expressed RanGAP1 proteins are localized in the cytoplasm during interphase (our unpublished data), we speculate that both the phosphorylation of RanGAP1 by CK2 and its complex formation with Ran and RanBP1 occur in the cytoplasm. Further studies will be required to more clearly understand whether nucleocytoplasmic transport activity is actually regulated through the phosphorylation of RanGAP1.
Given the fact that the acidic region of yeast Rna1p is involved in microtubule organization during mitosis, it is conceivable that the phosphorylation of the corresponding region of vertebrate RanGAP1 proteins may also play a role in mitosis. It has been reported that the targeting of RanGAP1 to the kinetochore and mitotic spindle is achieved by the binding of sumoylated RanGAP1 to RanBP2 (Joseph et al., 2004). The unsumoylated RanGAP1 protein, especially if it is in the active form, may exhibit deleterious effects on the formation of the Ran-GTP gradient during mitosis, unless it is correctly localized or inactivated. On the other hand, it has been shown that CK2 is associated with mitotic spindles (Litchfield, 2003). Therefore, the formation of a stable complex of phosphorylated RanGAP1, Ran and RanBP1 raises the possibility that stabilization of the ternary complex may accelerate the attachment of RanGAP1 to mitotic spindles (in this case RanBP2 may substitute for RanBP1) during mitosis. The formation of a stable complex may simultaneously inactivate excess RanGAP1 proteins. Indeed, unsumoylated RanGAP1, regardless of its origin (endogenous or over-expressed), appeared to be almost completely phosphorylated and only a fraction of the sumoylated RanGAP1 is not phosphorylated (Fig. 4B). In this context, it would be interesting to determine whether phosphorylation plays some role in Ran-GTP gradient formation during mitosis.
We wish to thank Dr. T. Nishimoto (Kyushu University) for the Ran cDNA, Dr. H. Saitoh (Kumamoto University) for the GST-SUMO97GG construct, and Dr. T. Tachibana (Osaka City University) for the RanGAP1 cDNA. This work was supported in part by grants-in-aid from the Japanese Ministry of Education, Culture, Sports, Science and Technology and the Human Frontiers Science Program.
|