| To whom correspondence should be addressed: Tamotsu Yoshimori, Department of Cell Genetics, National Institute of Genetics, Yata 1111 Mishima, Shizuoka 455-8540, Japan. Tel: +81–559–81–6881, Fax: +81–559–81–6884 E-mail: tamyoshi@lab.nig.ac.jp Abbreviations: PI3K, phosphatidylinositol 3-kinase; LPS, lipopolysaccharide; BSA, bovine serum albumin; GFP, green fluorescent protein; MβCD, methyl-β-cyclodextrin; CTxB, cholera toxin subunit B; DMEM, Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; RT, room temperature. |
Many bacterial pathogens are known to enter nonphagocytic cells (Cossart and Sansonetti, 2004), and two types of mechanism mediating bacterial entry into host cells have been extensively studied: trigger and zipper mechanisms (Finlay and Cossart, 1997; Lafont and van der Goot, 2005; Rottner et al., 2005). Salmonella typhimurium and Shigella flexneri deliver their virulence factors into the cellular cytoplasm with type III secretion system (TTSS), following their adherence to the target cells. These virulence factors elicit actin rearrangement, leading to entry triggered by the bacteria (trigger mechanism). In the zipper mechanism, the bacterial internalization is mediated by cellular actin rearrangement which is induced through specific interactions between bacterial ligands and cell surface receptors. For example, Listeria monocytogenes and Yersinia pseudotuberculosis express their specific adhesions, such as internalin and invasin for cellular components and extracellular matrix proteins, respectively, and these bindings activate the entry mechanisms. Rho family GTPases, such as Rac and Cdc42, are involved in actin rearrangement in both mechanisms (Gruenheid and Finlay, 2003). For several bacteria, lipid rafts in the plasma membrane have been suggested as bacterial entry sites (Garner et al., 2002; Lafont et al., 2002; Seveau et al., 2004; Zobiack et al., 2002). Lipid rafts are membrane subdomains rich in cholesterol, sphingolipids and specific membrane proteins, such as glycosylphosphatidylinositol-anchored proteins (GPI-APs). It has been suggested that lipid rafts act as platforms in protein sorting and signal transduction (Simons and Ikonen, 1997). In addition to these roles, lipid rafts may serve as entry sites for bacteria. Recently, Veiga and Cossart reported that Listeria monocytogenes hijacks the clathrin-mediated endocytic machinery for its entry into its host cells (Veiga and Cossart, 2005). However, the details of its mechanism still remain largely unknown.
Periodontitis is one of the most common chronic diseases afflicting mankind, and is characterized by the destruction of supporting periodontal connective tissue and tooth loss. The disease has been also implicated in atherosclerosis (Dorn et al., 1999; Gibson et al., 2004; Haraszthy et al., 2000) and pre-term delivery of low birth weight infants (Offenbacher et al., 1996). Porphyromonas gingivalis, a Gram-negative short rod anaerobe, has been identified as a bona fide pathogen of adult periodontitis (Socransky and Haffajee, 1992). P. gingivalis was detected within gingival tissues of periodontitis patients (Papapanou et al., 1994), and was also reportedly internalized by several human epithelial cell lines in vitro (Duncan et al., 1993; Lamont et al., 1995). Thus, those bacterial interactions with epithelial cells are considered to be the key events to establish chronic periodontal infection (Lamont and Yilmaz, 2002). P. gingivalis initiates its entry to host cells through the interaction between cellular α5β1 integrin molecules and fimbriae, which are filamentous appendages on the bacterial surfaces (Nakagawa et al., 2002; Yilmaz et al., 2002). However, the further molecular machinery involved in the event is unclear. A large number of Gram-negative bacteria, including P. gingivalis, have an ability to extracellularly release membrane vesicles (MVs). This takes place during normal growth, possibly as a result of cell wall turn over (Grenier and Mayrand, 1987; Mayrand and Grenier, 1989; Zhou et al., 1998). In fact, the MVs retain full components of outer membrane constituents of cell wall including proteins, LPS, muramic acid, capsule, and fimbriae (Zhou et al., 1998). MVs isolated from culture medium of P. gingivalis can be conjugated to polystyrene fluorescent-beads, and the beads were internalized by epithelial cells in a MVs-dependent manner (Inaba et al., 2006). Use of the MVs-coated beads as a homogenous artificial intruder allows us to quantify the internalization efficiency easily and stably, and would potentially avoid the complications arising from living bacterial organisms such as multiplication.
Here, we developed a new assay system to measure internalization of the MVs-coated beads to cultured cells, and used it to characterize the entry mechanisms of P. gingivalis with respect to the host cellular endocytic machinery, cytoskeleton, and lipid rafts.
All Alexa Fluor dyes-conjugated reagents were purchased from Molecular Probes Inc. (Eugene, OR, USA), EZ-link sulfo-NHS-LC-biotin was from Pierce (Rockford, IL, USA), mouse monoclonal anti-tubulin α antibody, cytochalasin D, nocodazole, taxol, methyl-β-cyclodextrin (MβCD), filipin III, and nystatin were from Sigma (St Louis, MO, USA), latrunculin A and wortmannin were from Wako (Osaka, Japan). The dominant-negative Eps15 construct GFP-Eps15Δ95/295, in which the second and third EH domains are deleted, was kindly provided by Dr. A. Dautry-Varsat (Institut Pasteur, Paris, France). Using this plasmid, we constructed GFP-Eps15ΔEH in which all of the three EH domains are lacking. GFP-dynamin 2 and GFP-dynamin 2-K44A were generous gifts from Dr. K. Nakayama (Kyoto University, Kyoto, Japan). Caveolin-1-GFP was a kind gift from Dr. A. Helenius (Swiss Federal Institute of Technology, Zurich, Switzerland).
HeLa cells were grown in DMEM (Sigma) supplemented with 10% FBS (Invitrogen, Carlsbad, CA), 4 mM L-glutamine (Invitrogen) and 10 μg/mL gentamicin (Sigma). 6×104 HeLa cells seeded on coverslips in 24-well plates were transiently transfected with 1.0 μg of the plasmid by LipofectAMINE 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s recommendations. Transfected cells were further incubated for 20 h and used for the assays. P. gingivalis TDC60 (type II fimA) was grown in GAM broth (Nissui, Tokyo, Japan) supplemented with 5 μg/ml hemin and 1 μg/ml menadione anaerobically (80% N2, 10% H2, 10% CO2).
P. gingivalis extracellular vesicles were prepared according to the Rosen method (Rosen et al., 1995). Briefly, 500 ml of cell culture was subjected to centrifugation at 10,000×g for 20 min. The culture supernatant was filtrated through a 0.2 μm-pore-size filter (Nalge Nunc, Rochester, NY), and centrifuged at 100,000×g for 50 min. The precipitates containing extracellular vesicles were suspended in 500 μl of PBS (~2.5 μg proteins/ml). For heat-inactivation, the MVs suspension was incubated at 70°C for 30 min.
Alexa 365- or 580-conjugated sulfate-modified polystyrene beads (1.0 μm in diameter) were used for the bead internalization assay. First, the beads were washed three times with PBS. The sulfate-modified beads (1.45×1010) were resuspended in PBS (50 μl) containing MVs (1 μg/ml), heat-inactivated MVs (1 μg/ml) or BSA (200 μg/ml), and incubated at room temperature (RT) for 4 h. The MVs- or BSA-coated beads were washed three times with PBS and resuspended in 1 ml of PBS containing 1% BSA.
8×104 HeLa cells were seeded onto glass coverslips in 24-well plates (Nalge Nunc) the day before the assay. The cells were washed twice with DMEM and incubated for 30 min in the presence or absence of indicated reagents. 1.35×107 of MVs-coated beads were added to the cells. The plate was centrifuged at 410×g for 1 min at 37°C to promote contact between the beads and the cells. After being incubated at 37°C for 1 h, the plate was transferred to 4°C. The cells were washed with ice-cold PBS and incubated with sulfo-NHS-LC-biotin (100 μg/ml in PBS) at 4°C for 15 min. Subsequently, the cells were washed twice and exposed to streptavidin-Alexa Fluor 488 (10 μg/ml in PBS) at 4°C for 10 min. Finally, the cells were washed and fixed with 3% paraformaldehyde (PFA) in PBS for 10 min.
An Olympus FV1000 laser scanning confocal microscope (Olympus, Tokyo, Japan) was used for image collection and analysis. The images were taken with a 100×objective oil immersion lens at 800×800 pixel resolution. To quantify the beads internalized by the cells, maximal projection images were made. In each field, intra- and extracellular beads were counted manually. At least 6 fields and ~200 cells were analyzed for each experiment, and three independent experiments were done. The results were normalized by the control experiments.
HeLa cells were washed twice with serum-free DMEM and incubated with the following drugs in DMEM at 37°C for 30 min prior to the assay. To disorganize the cytoskeletal architecture, 1 μg/ml cytochalasin D, 2 μM latrunculin A, 25 μM nocodazol or 50 μM taxol was used. To analyze the role of cholesterol in bead internalization, 10 mM MβCD, 5 μg/ml filipin III or 50 μg/ml nystatin was used. To inhibit PI3K activity, 100 nM wortmannin or 50 μM LY294002 was used. All reagents were included in the medium throughout the internalization experiments.
For immunostaining, cells were washed with ice-cold PBS, fixed with 3% PFA in PBS for 10 min, and then permeabilized with 0.1% Triton X-100 in PBS for 10 min. After being washed twice with PBS, the cells were incubated in blocking solution (0.1% gelatin in PBS) for 5 min and subsequently with primary antibodies diluted with the blocking solution at RT for 1 h. To stain F-actin, cells were incubated with 0.25 μM of Alexa 488-phalloidin in PBS/1% BSA at RT for 30 min. For GM1 labeling, cells were fixed with PFA and subsequently labeled with 40 μg/ml of Alexa 488-conjugated cholera toxin subunit B (Molecular Probes Inc.) for 30 min on ice. The cells were observed with an Olympus FV1000 laser scanning confocal microscope.
To precisely measure the internalization of MVs-coated beads to cells, we developed an assay as illustrated in Fig. 1A. HeLa cells were incubated with the MVs-coated, magenta-colored beads (1 μm in diameter) for 1 h, washed with PBS, and then the plasma membrane of the host cells as well as extracellular beads attaching cell surface were biotinylated with membrane impermeable biotin. Finally, Alexa 488 (green) conjugated streptavidin was added to cells to visualize the biotinylated molecules (see Materials and Methods). The result is shown in Fig. 1B. If the beads just adhered to the host cell surface or were incompletely internalized, they were accessible to the biotin-streptavidin labeling. As a result, they became white by color merge and were easily distinguished from completely internalized beads with the magenta image. The cell boundaries were also confirmed by the vertical optical sections. The MVs-coated beads were efficiently internalized by cells for 1 h. If the beads were coated with BSA or heat-inactivated MVs, they were hardly internalized (Fig. 1C), indicating that the internalization is dependent on the MVs. Fig. 1D shows time-dependent internalization of MVs-coated beads. The number of the completely internalized beads increased along the incubation time, and reached saturation at 40 min. The kinetics of internalization was found to be quite similar to that of living bacteria reported in previous studies (Yilmaz et al., 2004; Yilmaz et al., 2003).We also examined the localization of α5β1-integrin which was reported to be responsible for the initial binding of P. gingivalis to the cells (Nakagawa et al., 2005; Yilmaz et al., 2002). Consistent with those previous reports, recruitment of α5β1-integrin was observed at the bead internalization site (Fig. 1E).
 View Details | Fig. 1. MVs-coated beads are internalized by HeLa cells. (A) A scheme for surface biotinylation to distinguish internalized beads from extracellular ones. HeLa cells were incubated with the MVs-coated beads (magenta) for 1 h and subjected to biotinylation with sulfo-NHS-LC-biotin. Subsequently, the cells were treated with Alexa 488-conjugated streptavidin (green), and then fixed with paraformaldehyde. (B) In cells treated as in (A), confocal optical sections of 0.18 μm thickness were taken at 0.5 μm intervals. A projection image and vertical (z) optical sections (x-z and y-z planes) are shown. The magenta arrow indicates completely internalized beads. The white arrow indicates beads that just adhered to the host cell surface or were incompletely internalized. Bar, 4 μm. (C) Comparison of entry efficiency among MVs-, BSA- and heat inactivated MVs-coated beads. The number of the magenta-colored beads per cell was quantified. The mean values from three independent experiments are shown with SEs. (D) Time-dependent internalization of the MVs-coated beads. At the beginning of the assay, the cells were incubated with the MVs-coated beads for the indicated time periods. (E) Recruitment of α5β1-integrin to the entry of MVs-coated bead. After 1 h of incubation with the beads, HeLa cells were processed for the anti-α5β1-integrin antibody staining. The boxed area shows that α5β1-integrin (magenta) is recruited around the MVs-coated bead (cyan). Bar, 5 μm. |
Eps15 is required for clathrin-dependent endocytosis. This protein contains three EH (Eps15-homology) domains, and overexpression of the Eps15 mutant lacking the EH domains interferes with clathrin-coated pit assembly (Benmerah et al., 1999). To examine whether the MVs-coated beads are internalized by cells in a clathrin-dependent manner, GFP-Eps15ΔEH was transiently transfected to HeLa cells. As expected, uptake of transferrin, a marker for clathrin-dependent endocytosis, was inhibited in the transfected cells (Fig. 2A). In contrast, the transfected cells were found to internalize the MVs-coated beads (Fig. 2B), indicating that the bead internalization is a clathrin-independent process. Next, we analyzed the involvement of the GTPase dynamin in bead internalization. The dynamin family contains three members Dyn1, 2 and 3, with Dyn2 being a ubiquitously expressed form (Cao et al., 1998). GTPase activity of Dyn2 is required for both clathrin-dependent and -independent endocytosis (Lamaze et al., 2001; Le and Nabi, 2003; Oh et al., 1998; Sabharanjak et al., 2002), and the mutant Dyn2-K44A defective in GTP-binding shows a dominant negative effect. As shown in Fig. 2C, the beads were internalized by the cells overexpressing the wild type Dyn2 fused with GFP. In contrast, overexpression of the dominant negative GFP-Dyn2-K44A drastically prevented bead internalization (Fig. 2D). Notably, the beads were aggregated on the cell surface, and GFP-Dyn2-K44A was apparently clustered together with the beads. These results indicate that the beads are internalized by the cells in a clathrin-independent and dynamin-dependent manner.
 View Details | Fig. 2. Overexpression of the dominant-negative mutant dynamin 2 inhibits the internalization of MVs-coated beads but the Eps15 mutant does not. HeLa cells transfected with GFP-tagged Eps15ΔEH (A, B), GFP-Dyn2-WT (C) or GFP-Dyn2-K44A (D) were incubated with Alexa568-conjugated transferrin (A: magenta) or MVs-coated beads (B, C, D: magenta) at 37°C for 1h, washed, fixed and permeabilized. Cells were then stained with DAPI (B, D: cyan). A projection image at mid-height of the cell and vertical (z) optical sections (x-z and y-z planes) are shown (GFP: green). White outlines show perimeters of non-transfected cells. Arrows in (B) and (C) indicate the internalized beads, arrowheads in (D) indicate recruitment of GFP-Dyn2-K44A to the aggregated beads on the cell surface. Bars, 10 μm. |
To evaluate the role of the actin cytoskeleton in the bead internalization, the internalization assay was carried out in the presence of the actin polymerization inhibitors, cytochalasin D and latrunculin A. As shown in Fig. 3A and B, bead internalization was significantly prevented by these reagents. Almost all the beads in the images were white, indicating that they just adhered to the cell surface or were incompletely internalized. Next, we examined the effect of microtubule disorganization on bead internalization, using the microtubule assembly inhibitor nocodazole and the microtubule-stabilizing drug taxol. These drugs also inhibited the bead internalization by more than 65% (Fig. 3A and B). The effect of taxol indicates that not only microtubule assembly but also disassembly is critical to the process. To investigate whether actin filaments and microtubles are remodeled by the MVs-coated bead internalization, the cells were stained with phalloidin for actin and the anti-α-tubulin antibody for microtubules. These staining images revealed that some beads were surrounded by both F-actin and α-tubulin (Fig. 3C, arrowheads). Although it is unclear whether the internalization of the surrounded beads was ongoing or just finished, the vertical optical sections (the x-z plane) indicate that both actin cytoskeleton and microtubules function in the uptake pathway. It is likely that the actin remodeling, which is required for engulfment of the MVs-coated beads, is induced by attachment of the beads to the plasma membrane. Interestingly, when the cells were treated with either nocodazole or taxol, the recruitment of F-actin around the beads was not abolished (Fig. 3D), suggesting that the recruitment of α-tubulin follows the engulfment of beads by F-actin.
 View Details | Fig. 3. Internalization of the MVs-coated beads is dependent on both actin and microtubule cytoskeletons. The bead internalization assay described in Fig. 1A and Materials and Methods was carried out in the presence of the indicated drugs. (A) Each image is a projection of 4–5 confocal sections of the middle region (beads: magenta, surface label: green). Bars, 20 μm. (B) The number of the magenta-colored beads per cell was quantified, and presented as percentage of control cells, which were not treated with the drugs. The mean values from three independent experiments are shown with SEs. (C) After 40 min of incubation with the MVs-coated beads (cyan), the cells were processed for staining with anti-α-tubulin (green) and Alexa Fluor 568-conjugated phalloidin (magenta). Images of x-y and x-z planes are shown. Arrowheads indicate colocalization of beads with both F-actin and α-tubulin. Arrow indicates colocalization of beads with F-actin. (D) After incubation with the MVs-coated beads in the presence of taxol (upper panels) or nocodazole (lower panels), cells were stained as described in (C). Insets show enlarged images of the boxed areas. Bars, 5 μm. |
Phosphatidylinositol 3-kinase (PI3K) is involved in rapid actin polymerization during phagocytosis in macrophages (Cox et al., 1999). To elucidate whether the PI3K activity contributes to bead internalization, we examined the effect of PI3K inhibitors, wortmannin and LY294002. As shown in Fig. 4A and B, these drugs strongly prevented bead internalization, which indicates that PI3K is involved in bead internalization.
 View Details | Fig. 4. Internalization of the MVs-coated beads is dependent on PI3K. (A) The bead internalization assay described in Fig. 1A and Materials and Methods was carried out in the presence of the PI3K inhibitors, wortmannin and LY294002. Each image is a projection of 4–5 confocal sections of the middle region. Bars, 10 μm. (B) The number of the internalized beads (magenta) per cell was quantified, and presented as percentage of control cells, which were not treated with drugs. The mean values from three independent experiments are shown with SEs. |
Recently, accumulating evidence has revealed that lipid rafts serve as entry sites into host cells for some bacteria (Lafont et al., 2004). To examine whether this is the case with P. gingivalis, we perturbed the raft formation by treatment with filipin, nystatin, or methyl-β-cyclodextrin (MβCD). Filipin and nystatin bind to and sequester cholesterol, an essential component of lipid rafts, while MβCD removes cholesterol from the membrane (Kilsdonk et al., 1995; Smart and Anderson, 2002). As shown in Fig. 5A, the number of magenta-colored beads, which were completely internalized by the cells, significantly decreased by these drugs, i.e., 40~50% inhibition by filipin and nystatin, and 95% inhibition by MβCD. (Fig. 5B). These results indicate that cholesterol is required for internalization of the MVs-coated beads.
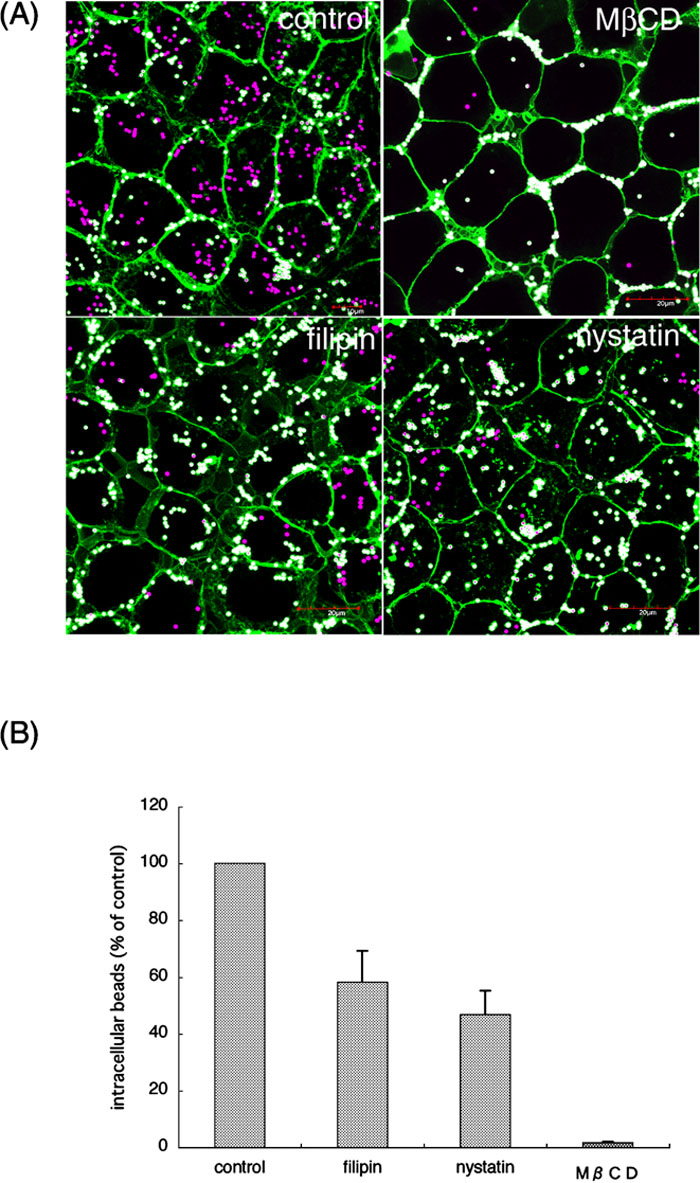 View Details | Fig. 5. Internalization of the MVs-coated beads is dependent on cholesterol. (A) The bead internalization assay described in Fig. 1A and Materials and Methods was carried out in the presence of the indicated drugs. Each image is a projection of 4–5 confocal sections of the middle region. (B) The number of the internalized beads (magenta) per cell was counted, and presented as percentage of control cells, which were not treated with the drugs. The mean values from three independent experiments are shown with SEs. |
Next, we investigated colocalization of raft markers, ganglioside GM1 and caveolin-1, with the MVs-coated beads. After being incubated with the beads, cells were fixed and cell surface GM1 was labeled with fluorescent cholera toxin subunit B (CTxB). As shown in Fig. 6A, GM1 was clearly recruited around the beads. In addition, the beads were surrounded by caveolin-1-GFP (Fig. 6B). Thus, lipid rafts were shown to be indeed localized to the beads entry site.
 View Details | Fig. 6. Recruitment of raft markers to the MVs-coated beads. (A) HeLa cells were incubated with the MVs-coated beads (magenta), washed and fixed. The cells were then incubated with Alexa 488-conjugated CTxB to label GM1. (B) HeLa cells transiently expressing caveolin-1-GFP were incubated with the MVs-coated beads (magenta), washed and fixed. Recruitment of caveolin-1 (green) around the beads is indicated in boxed areas. Insets show enlarged images of the boxed areas. Bars, 5 μm. |
In this study, we have studied the contribution of several host cell factors to P. gingivalis internalization, using fluorescent beads coated with the MVs released by this bacterium. Combined use of the MVs-coated beads and cell surface-biotinylation provided an easy and reliable assay system for the internalization to cells. The MVs-coated beads were efficiently internalized by nonphagocytic HeLa cells, indicating that the bacterial components of the outer membrane are sufficient for the internalization. The present internalization mode is apparently distinct from the trigger mechanism, because P. gingivalis does not produce TTSS (Lamont and Yilmaz, 2002). Previous studies of living P. gingivalis (Deshpande et al., 1998; Lamont et al., 1995; Nakagawa et al., 2005; Yilmaz et al., 2002) were reminiscent of the zipper mechanism. Those previous findings are evidenced by the present study showing that actin cytoskeleton and PI3K are required for the internalization and that α5β1-integrin, a receptor for the bacterium, and F-actin were localized around the beads.
We further demonstrated novel cellular players involved in the internalization of P. gingivalis to cells. We found that the internalization of the MVs-coated beads is dependent on Dyn2. Although Dyn2 is required for scission of the clathrin-coated vesicles, it is also involved in clathrin-independent processes, such as caveolar endocytosis (Nichols, 2003) and phagocytosis in macrophages (Gold et al., 1999). It has been recently suggested that Dyn2 regulates actin polymerization at the site of plasma membrane invagination, in both clathrin-dependent and -independent internalization (Sauvonnet et al., 2005). This is probably achieved by binding of dynamin to F-actin-interacting proteins (Orth and McNiven, 2003; Schafer, 2004). Therefore, we propose that actin dynamics controlled by Dyn2 is critical for internalization of P. gingivalis. In contrast to Listeria monocytogenes (Veiga and Cossart, 2005), Eps15 was dispensable for internalization of P. gingivalis.
We also suggested for the first time that dynamics of microtubule assembly and disassembly are required for the internalization of P. gingivalis. It is known that microtubules are involved in entry of several pathogens into epithelial cells (Yoshida and Sasakawa, 2003). Nocodazole inhibited the entry in most of the cases, whereas taxol did not (Grassme et al., 1996; Kuhn, 1998; Oelschlaeger et al., 1993). It is known that taxol does not inhibit motor-dependent processes. Internalizations of Campylobacter jejuni (Hu and Kopecko, 1999) and Chlamydia trachomatis (Clausen et al., 1997), both of which require a dynein motor, were not prevented by taxol. On the contrary, the MVs-coated bead internalization was inhibited by both nocodazole and taxol. This suggests that the internalization does not require stable microtubules as a rail for the motor-dependent sliding, and rather that it needs dynamic polymerization and depolymerization of microtubules. Tubulin localized around the beads together with F-actin. Considering that neither nocodazole nor taxol inhibited the formation of F-actin-rich region around the beads, microtubule assembly and disassembly around the beads may follow the step of actin polymerization to form the membrane-bound compartment engulfing the beads.
Recently, it was reported that siRNA-mediated reduction of caveolin-1 expression caused inhibition of P. gingivalis internalization, but the colocalization of intracellular P. gingivalis with caveolin-1 was not clearly shown (Tamai et al., 2005). In addition, the inhibitory effect of MβCD for the internalization was reportedly only 50%. Judging from those observations, the involvement of lipid rafts in the internalization cannot be said to be conclusive. However, in our experimental system, MβCD was found to be a significant inhibitor for the bead internalization. We also showed an inhibitory effect of nystatin and filipin. Further, the intracellular beads were apparently colocalized with lipid raft markers (Fig. 6). These results strongly suggest that lipid rafts play a pivotal role in the internalization process of P. gingivalis, and that cholesterol is required for formation of the lipid rafts serving as the putative bacterial entry sites. However, more detailed analysis would be required to substantiate the hypothesis.
In summary, we developed a novel assay method to reliably and conveniently examine P. gingivalis internalization to cells, and applied it successfully to obtain important insights into the molecular basis underlying the bacterial entry to host cells. Internalization of the MVs-coated beads requires host cellular dynamin, actin fibers, microtubules, PI3K, and the lipid rafts. Such a combination is so far unknown for other bacterial entry, indicating the particularity of P. gingivalis internalization to cells and suggesting a diversity of bacterial entry mechanisms. To our surprise, the dynamics of microtubule assembly and disassembly is essential to the process, whereas other bacterial entry mechanisms usually employ stable microtubules. Future studies should unravel how the microtubule dynamics is involved in the process, what is the role of the lipid rafts, and what relationship exists among each of the components, that is, dynamin, actin, microtubules, and the lipid rafts. The present bead internalization assay method will no doubt be useful to further investigate these issues.
We wish to thank Dr. K. Nakayama (Kyoto University) for the dynamin 2 constructs, Dr. A. Dautry-Varsat (Institut Pasteur) for the Eps15 constructs, and Dr. A. Helenius (Swiss Federal Institute of Technology) for caveolin-1-GFP. We are also very grateful to Dr. N. Shiina (National Institute of Genetics) for helpful discussions. This work was supported in part by Special Coordination Funds for Promoting Science and Technology of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT).
|