| To whom correspondence should be addressed: Takahiko Yokoyama, MD, Department of Anatomy and Developmental Biology, Kyoto Prefectural University of Medicine, Kawaramachi-Hirokoji, Kamigyo-ku, Kyoto 602-0841, Japan. Tel: +81–75–251–5303, Fax: +81–75–251–5304 E-mail: tyoko@koto.kpu-m.ac.jp |
Monocilia (primary cilia) used to be considered a vestigial or remnant structure of no functional importance. However, recent studies have shown that primary cilia are important in the establishment of body left-right asymmetry and to maintain normal renal tubular architecture. During early developmental stages, primary cilia in the node are motile and create leftward fluid flow by rapidly rotating themselves. Studies of nodal primary cilia are performed in mutants that show randomization of body situs, such as kif3a, kif3b, Tg737, iv and pkd2 mutant mice (Supp et al., 1997; Nonaka et al., 1998; Takeda et al., 1999; Murcia et al., 2000; Pennekamp et al., 2002). kif3a, kif3b and Tg737 mutants fail to produce nodal primary cilia. iv and pkd2 mutants possess primary cilia, but iv mutant cilia are immotile (Okada et al., 1999). The pkd2 mutant lacks a Ca2+ response in the node during development (McGrath et al., 2003). In addition to randomization of body situs, kif3a, Tg737 and pkd2 mutants develop multiple renal cysts (Moyer et al., 1994; Wu et al., 2000; Lin et al., 2003). In contrast to nodal motile cilia, primary cilia in renal epithelial cells are non-motile. Renal epithelial cells of kif3a and Tg737 mutants show a loss or shortened cilia in vivo and in vitro (Pazour et al., 2000; Yoder et al., 2002; Lin et al., 2003). Renal cells derived from the pkd1 mutant or cells treated with polycystin2 (a gene product of pkd2) antibody fail to increase intracellular Ca2+ concentrations in response to fluid stress (Nauli et al., 2003). Furthermore, isolated renal tubules in Tg737 mutants displayed blunted increases in intracellular Ca2+ concentration in response to fluid stress compared to normal tubules (Liu et al., 2005). Thus, abnormal structures in primary cilia and/or impairments in increases in intracellular Ca2+ concentration in response to fluid flow are thought to cause renal cyst formations.
One explanation why loss or truncation of cilia causes renal cyst formation is that non-motile primary cilia on renal epithelial cells function as a flow sensor (Praetorius and Spring, 2003; Yokoyama, 2004). Fluid flow can bend primary cilia of rat kangaroo cells (PtK1) (Schwartz et al., 1997). Bending a cilium by pipette or fluid flow induced Ca2+ influx in Madin-Darby canine kidney cells (MDCK) (Praetorius and Spring, 2001). Nauli et al. showed that fluid flow increased intracellular Ca2+ in collecting tubule cells derived from normal mice, but not from pkd1 null mice or renal cells treated with anti-pkd1/2 protein antibodies (Nauli et al., 2003). Taken together with a recent report concerning abundant cation-permeable channel activities in the ciliary membrane (Raychowdhury et al., 2005), it is hypothesized that the pkd1/pkd2 complex could function as a molecular sensor as well as a Ca2+ channel, and that a lack of flow-sensing in primary cilia could lead to renal cyst formation.
The inv (inversion of embryonic turning) mouse mutant was discovered in a family of transgenic mice that showed situs inversus associated with multiple renal cysts (Yokoyama et al., 1993; Mochizuki et al., 1998). Mutation in the inv gene in human was later found to cause nephronophthisis type 2 (NPHP2) (Otto et al., 2003). Recently, primary cilia in the primitive node of inv/inv mutants were reported to show aberrant rotation and subsequently made turbulent nodal flow (Okada et al., 1999, 2005). Artificial leftward nodal flow rescued situs inversus in inv/inv mutants in vitro, suggesting that the turbulent nodal flow causes situs abnormalities (Watanabe et al., 2003). It is possible that a dysfunction of the primary cilia machinery is responsible for the turbulent nodal flow. Scanning electron microscopy (SEM) analysis of inv/inv kidney sections showed normal appearing primary cilia at cystic tubules (Phillips et al., 2004), but response of primary cilia in inv/inv mutants to fluid stress has yet to be clarified.
In the present study, we first examined subcellular localization of inv proteins and whether flow stimulation affects localization of inv protein. The main purpose of the present study was to examine if inv/inv mutant renal cells have any abnormalities in mechanical response of primary cilia to physiological fluid flow, or abnormalities in intracellular Ca2+ increase in response to fluid stress.
One-μm-diameter polystyrene beads were purchased from Polysciences, Inc. (Warrington, PA). Fura-2 AM and Pluronic F127 gel were from Molecular Probes (Eugene, OR). Fluorescein-conjugated LTA (LTA-FITC) was obtained from Vector Laboratories (Burlingame, CA). Cell culture supplements were obtained from Invitrogen (Carlsbad, CA). Unless otherwise stated, all chemicals were purchased from Sigma (St Louis, MO) or Wako Pure Chemical (Osaka, Japan).
Normal, inv/inv and inv-GFP transgenic mice (Watanabe et al., 2003) were maintained in an animal facility according to experimental procedures that were approved by the Committee for Animal Research, Kyoto Prefectural University of Medicine. Mice (postnatal day 5) were anesthetized by intraperitoneal administration of sodium pentobarbital at a dose of 50 mg/kg body weight. Kidneys were isolated and dissociated with Krebs buffer containing 10% BSA and 1 mg/ml collagenase for 30 min with gentle shaking at 37°C. Digested tissue fragments were passed through 125 μm, 105 μm and 45 μm sieves, and centrifuged at 1000×g for 10 min at room temperature. The pellet was resuspended in Dulbecco’s modified Eagle’s medium/F-12 medium containing 10% fetal bovine serum, and cells were seeded on plastic dishes or glass coverslips coated with human collagen IV (50 μg/ml). Cells were incubated at 37°C, and equilibrated with 5% CO2 in humidified air. After 24 h of incubation, culture medium was changed to D-MEM / F-12 medium containing 0.5% fetal bovine serum, 100 μM MEM non-essential amino acid solution, 5 mg/l insulin, 5 μg/l sodium selenite, 5 mg/ml transferrin, 400 μg/l dexamethasone, 10 ng/ml epidermal growth factor, 5 pg/ml 2,3,5-triido-l-thyronine, 10000 U/l penicillin, 100 mg/l streptomycin, and 250 μg/l Fungizone®. Medium was changed daily.
Primary renal epithelial cells were grown on type IV collagen-coated glass coverslips for at least 2 days. Cells were placed in a parallel plate-type perfusion chamber (FSC2 closed system, Bioptechs, Butler, PA). The flow chamber was set on the stage of an inverted microscope (IX70, Olympus, Tokyo, Japan) equipped with a CCD camera (UIC-QE, Molecular Devices Corporation, Sunnyvale, CA). One end of the chamber was connected to a reservoir filled with Hanks balanced salt solution via a silicon tube. The chamber and reservoir were maintained at 37°C by a temperature sensor and heater (FCS2 controller, Bioptechs). Fluid flow was applied to cells by adjusting the height of the reservoir, and averaged volume flow (ml/s) was calculated from changes in weight of the reservoir. We captured Nomarski images of primary cilia using MetaFluor (Molecular Devices Corp., Sunnyvale, CA) for Windows every 25 msec. One μm-diameter polystyrene beads were used for determining linear fluid velocity profiles at the level of primary cilia. When averaged linear velocity in the chamber was 3.1 mm/s, linear fluid velocity at the level of the primary cilia (at 10 μm) was about 280 μm/s under our experimental conditions (Fig. 1A). Linear fluid velocity applied in the present study corresponded to an appropriate physiological range of proximal tubular flow rates (Chou and Marsh, 1987). Percentage of ciliated cells was assessed by microscope. Length of primary cilia (L, μm) was determined as follows:
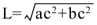 | (Fig. 1B). |
 View Details | Fig. 1. (A) Analysis of flow velocity and (B) primary cilium length. (A) Fluid velocities were measured by tracking the displacement of 1-μm polystyrene beads. At least 10 beads were used to characterize fluid velocities. Representative flow profiles are shown. The velocity profile between the two planes is parabolic. However, the velocity profile in a small distance from the cell surface (about 30 um) became linear. (B) Schematic diagram of a primary cilium. Plane α is the apical cell surface. Length of primary cilia (L, μm) was determined as follows: a: the top of the cilium, b: the base of the cilium, c: a point where cell surface and a vertical line from the “a” to apical cell surface cross. ac is the height of primary cilium (μm). Since the cilium was slightly tilted, the height does not correspond to the length of the cilium. The height (the distance between a and c) is determined by focusing from the base of the cilium (b) to the top of cilium (a). |
a: the top of the cilium, b: the base of the cilium, c: a point where the apical cell surface and a vertical line from the “a” to the apical cell surface cross. ac is the height of primary cilia (μm). Since the cilium was slightly tilted, the height does not correspond to the length of the cilium. The height (distance between a and c) was determined by focusing from the apical cell surface (b) to the top of cilia (a). (n>30).
Primary kidney epithelial cells derived from transgenic inv/inv mice expressing the inv-GFP transgene were grown on type IV collagen-coated glass coverslips for at least 2 days. GFP fluorescent images were obtained with an Olympus microscope (IX70) and a CCD camera (UIC-QE). Primary cilia were bent by fluid flow to make their entire length visible.
Primary renal epithelial cells were grown on type IV collagen-coated glass coverslips for at least 2 days. Cells were incubated for 30 min with the Ca2+ sensitive probe Fura-2 AM (at a final concentration of 5 μM) and 0.01% Pluronic F127 at room temperature, in serum free medium. Cells were washed twice to remove excess Fura-2 AM, and incubated for 15 min at 37°C for de-esterization. During de-esterization, cells were co-incubated with LTA-FITC (diluted 1:1000) for identification of proximal convoluted tubules (Laitinen et al., 1987). Subsequently, cells were placed in the chamber described above, and fluid flow was applied to the cells. Paired fluorescent images were captured using MetaFluor every 5 s at excitation wavelengths of 340 nm and 380 nm with a xenon light source. The fluorescent ratio (F340/F380) was monitored as changes in intracellular Ca2+ concentrations. Data were obtained from 7 regions of 2 to 5 cells.
Data are expressed as mean±S.E. Data obtained from the two groups were compared using a t-test. P values of less than 0.05 were considered significant.
Primary cilia of normal mice were seen as dots at the static state under Nomarski observation, suggesting that primary cilia extended perpendicularly to the apical membrane, and became parallel to the optical axis of the microscope (indicated by arrows in Fig. 2B). When physiological fluid flow (flow profiles in Fig. 1A) was applied, primary cilia bend and easily visible as lines (indicated by dashed circles in Fig. 2C). Successive Z-axis pictures of primary cilia both static and under fluid flow are available in the Supplementary Information, Video S1. In confluent cultures, ciliated cells were 81.3±1.3% in normal cells, and 78.2±3.7% in inv/inv mutant cells (Table I). Primary cilia were 11.5±0.6 μm in length in normal and 13.0±0.6 μm in inv/inv mutant cells (Table I). The percentage of ciliated cells and lengths of primary cilia were not significantly different between normal and inv/inv mutant cells (P>0.05).
 View Details | Fig. 2. Observation of primary cilia in normal mice-derived cells. Normal mice-derived primary kidney epithelial cells were cultured, and primary cilia were observed. Nomarski images at the level of cell nuclei (A), and at the level of primary cilia (B, C) are shown. Primary cilia were easily observed under fluid flow. Corresponding successive Z-axis pictures of primary cilia both at static and under fluid flow are available in the Supplementary Information, Video S1. Scale bars=10 μm. |
We examined the subcellular localization of functional inv protein in primary renal epithelial cells using transgenic inv/inv mice expressing the inv-GFP transgene, which rescues the complete phenotype of inv/inv mice, including kidney cyst formation and situs inversus. Fig. 3 shows Nomarski images at the level of cells/primary cilia (Fig. 3A to E) and corresponding inv-GFP fluorescent images (Fig. 3F to J) in primary cultured renal epithelial cells. Primary cilia were not observed as dots at the level of cell nuclei (Fig. 3A and D) and we could not detect inv-GFP fluorescence in the nucleus or in the membranes between cells (Fig. 3F and I). Primary cilia were clearly seen as dots above the cell nucleus level (Fig. 3B and C). We detected strong inv-GFP fluorescence in the base of primary cilia (Fig. 3G), but not in the top (Fig. 3H). Fluid flow was applied to visualize primary cilia fully (Fig. 3E), and the corresponding inv-GFP image showed strong GFP fluorescence in the base of primary cilia (Fig. 3J). Furthermore, the inv-GFP signal in the base of cilia did not translocate or change in intensity by physiological fluid flow for 90 min or more (data not shown).
 View Details | Fig. 3. Localization of functional inv protein to the base of primary cilia of kidney epithelial cells. inv/inv, inv-GFP mouse primary cilia of primary kidney epithelial cells. Images at the level of cell nuclei (A, D, F and I), and at the level of primary cilia (B, C, E, G, H and J) are shown. GFP fluorescence is detected at the base of primary cilia (G and J). Black arrows indicate the direction of fluid flow. Scale bars=10 μm. |
We analyzed the response of renal cilia to physiological flow stress. As shown in Fig. 2, primary cilia were observed as dots at the static state. When physiological fluid flow was applied, primary cilia of normal mice were bent. As soon as flow stimulation stopped, primary cilia quickly returned to their previous position. Fig. 4 shows consecutive pictures of a bending cilium every 25 msec when fluid flow stress was applied. It took on average 131±10 msec (n=10) from the beginning of bending to the completely bended state (Fig. 4A). When flow stress was stopped, cilia returned to the static state within an average time of 160±21 msec (n=10) (Fig. 4B). See the Supplementary Information, Video S2.
 View Details | Fig. 4. Primary cilium bending mechanics in response to fluid flow in normal mice-derived cells. Normal mice-derived primary kidney epithelial cells were cultured, and primary cilia were visualized. Representative consecutive pictures of a bending cilium at every 25 msec under fluid flow stress are shown. A) From the beginning of bending to the completely bended state. Flow is leftward. B) Primary cilia returned to the static state after the flow has stopped. Corresponding time-lapse video images (40 frames per second) are available in the Supplementary Information, Video S2. Scale bars=5 μm. |
Next, we examined whether inv/inv mouse cilia showed any abnormality in bending mechanics in response to flow stress. The inv/inv mouse cilia showed the same bending mechanics in response to physiological fluid flow as that of normal mouse cilia (Fig. 5). In inv/inv renal epithelial cells, it took on average 133±11 msec (n=10) from the beginning of bending to the completely bended state (Fig. 5A). When fluid flow was stopped, cilia returned to the static state within an average time of 160±14 msec (n=10) (Fig. 5B). See the Supplementary Information, Video S3. No statistical difference in bending and reflecting time of primary cilia between normal and inv/inv mutant cells was observed (P>0.05).
 View Details | Fig. 5. Primary cilium bending mechanics in response to fluid flow in inv/inv mice-derived cells. inv/inv mice-derived primary kidney epithelial cells were cultured, and primary cilia were visualized. Representative consecutive pictures of a bending cilium at every 25 msec under fluid flow stress are shown. A) From the beginning of bending to the completely bended state. Flow is leftward. B) Primary cilia returned to the static state after the flow has stopped. Corresponding time-lapse video images (40 frames per second) are available in the Supplementary Information, Video S3. Scale bars=5 μm. |
Cells were loaded with the Ca2+ indicator Fura-2. We selected LTA-positive proximal renal epithelial cells from both normal and inv/inv mutant mice to examine intracellular Ca2+ response to fluid flow (Fig. 6A and B). We detected a rise in intracellular Ca2+ concentration in response to fluid flow and this increase of intracellular Ca2+ concentration was maintained while fluid flow was applied. In normal cells, it took on average 96.4±4.7 sec to reach peak Ca2+ levels from the start of fluid stress. Ca2+ levels were maintained at higher than basal levels during fluid stimulation. After the flow was stopped, intracellular Ca2+ concentrations decreased and returned to basal levels within an average time of 153.6±15.3 sec.
 View Details | Fig. 6. Flow-induced Ca2+ responses in lectin-labeled, proximal convoluted tubule cells. Epithelial cells of proximal convoluted tubule origin were detected using LTA-FITC as markers. Cells loaded with Fura2-AM were exposed to fluid flow. Flow-induced Ca2+ responses in normal (A) and inv/inv (B) cells were analyzed. Representative data are shown. Detailed procedures are described in ‘Materials and Methods’. |
Inv/inv mutant cells also showed a rise in intracellular Ca2+ concentration in response to fluid flow and this increase of intracellular Ca2+ concentration was maintained while fluid flow was applied. inv/inv mutant cells took on average 99.9±4.9 sec to reach peak Ca2+ levels while fluid stress was applied. Ca2+ levels were maintained at higher than basal levels during fluid stimulation. After the flow was stopped, intracellular Ca2+ concentrations decreased, and returned to basal levels within an average time of 122.9±7.6 sec. There were no statistical differences between primary cilia of normal and inv/inv mutant cells in the time to reach the peak and the time to return to basal levels.
The present study provides three findings about renal cells of normal and inv/inv mice in response to fluid flow. First, functional inv protein was localized at the base of primary cilia and remained there even under fluid flow stimulation. Second, primary cilia of primary inv/inv mutant mouse renal epithelial cells bend in response to physiological fluid flow in an identical manner as those of normal mouse renal epithelial cells. Third, renal cells derived from inv/inv mice increased their intracellular Ca2+ concentration in response to physiological fluid flow.
Localization of inv protein has been reported to occur in cell membrane (Nurnberger et al., 2002; Simons et al., 2005), cytoplasm (Simons et al., 2005), nucleus (Nurnberger et al., 2002) and cilia (Morgan et al., 2002a; Otto et al., 2003; Watanabe et al., 2003). Previous reports except Watanabe et al. used antibodies against inv protein and cultured renal cell lines. Localization of inv protein using antibodies indicates the place where inv protein exists, but does not determine the place where the inv protein is functioning. The inv-GFP fusion construct rescues all the inv phenotypes. Thus, localization of the fusion protein indicates the place where the protein is functioning. In the present study, we showed that a strong GFP signal was observed at the base of primary cilia of primary cultured renal cells derived from inv-GFP mice at static state, and no translocation of the protein was observed after fluid flow stress, suggesting that the base of primary cilia is where the inv protein functions.
Primary cilia of mouse primary cultured renal epithelial cells stood straight and never displayed active beating under static conditions. In response to physiological fluid flow, primary cilia were bent, hence were easily visualized. As soon as the fluid flow stopped, primary cilia were able to return to their previous position without overshooting. These results correlated well with a previous report using renal cell lines of rat kangaroo (PtK1 cells) (Schwartz et al., 1997). Mutations in kif3a and Tg737 caused structural abnormalities of renal primary cilia in vivo and in vitro (Pazour et al., 2000; Yoder et al., 2002; Lin et al., 2003). Recently, primary cilia in the primitive node of inv/inv mutants were reported to show aberrant rotation and subsequently produced turbulent nodal flow, suggesting the possibility of a structural or functional alteration of primary cilia in inv mutant mice (Okada et al., 1999, 2005). However, our study showed that the lengths of primary cilia were almost identical in both normal and inv/inv mutant mice, and that irregularities in bending-and-return mechanics of inv/inv primary cilia were not observed under physiological fluid flow. Furthermore, ten times faster fluid flow did not eliminate primary cilium from the cell, indicating that inv/inv primary cilia are also firmly anchored to the cell (data not shown). Together with a previous SEM study (Phillips et al., 2004), it is unlikely that renal primary cilia in inv/inv mutants have structural abnormalities that cause renal cyst formation. Bending primary cilia in MDCK was reported to increase intracellular Ca2+ concentrations (Praetorius and Spring, 2001). Renal cells of pkd1 mutants or cells treated with anti-polycystin2 were unable to increase their intracellular Ca2+ concentration in response to physiological flow stress (Nauli et al., 2003). However, inv/inv cells showed intracellular Ca2+ increases after physiological flow stress that could bend primary cilia of renal cells the same way as normal cells. Although we cannot deny that more subtle difference may exist between normal and inv cells in the response or resting level of Ca2+, the present results strongly suggested that inv renal cells have the same Ca2+ response mechanism to flow stress as normal renal cells have.
Inv protein contains calmodulin-binding motifs, and Ca2+ controls calmodulin-inv binding (Yasuhiko et al., 2001; Morgan et al., 2002b). The polycystin complex acts as a Ca2+ channel (Hanaoka et al., 2000). Both inv protein and polycystin are localized in cilia. Losses of inv protein and polycystin-2 function lead not only to cyst formation, but also to situs inversus (Yokoyama et al., 1993; Pennekamp et al., 2002). Thus, there is a possible relationship between inv and the polycystin signaling pathway. Importantly, when mutant cells that lack inv were exposed to fluid flow, we detected Ca2+ influx. The present results suggest that inv protein participates in downstream signaling of Ca2+ influx. Recently, inv protein was shown to act on the Wnt pathway (Simons et al., 2005). It would be interesting to investigate whether polycystins also modulate the Wnt signaling pathway, and share a common pathway with inv.
In summary, inv renal cells show no structural abnormalities of cilia, and intracellular Ca2+ increases in response to physiological fluid flow are the same as in normal renal cells. Although the inv protein is localized in the cilia like polaris, kif3 and polycystins, the present results suggest that the inv protein has a distinct function.
This research was partially supported by the Mitsubishi Foundation and by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture (15370095) to T.Y. and for Young Scientists (17790142) to D.S. We are grateful to Dr. Hiroshi Hamada (Developmental Genetics Group, Graduate School of Frontier Biosciences, Osaka University) for providing inv/inv mice expressing the inv-GFP. We thank Drs. Joji Ando, Kimiko Yamamoto (Dept. of Biomedical Engineering, Graduate School of Medicine, University of Tokyo) and Hideo Tanaka (Department of Pathology and Cell Regulation, Graduate School of Medical Science, Kyoto Prefectural University of Medicine) for their valuable suggestions about mechanisms of flow-induced Ca2+ influx.
|