| To whom correspondence should be addressed: Ryo Iwamoto, Department of Cell Biology, Research Institute for Microbial Diseases, Osaka University, Suita, Osaka 565-0871, Japan. Tel: +81–6–6879–8288, Fax: +81–6–6879–8289 E-mail: riwamoto@biken.osaka-u.ac.jp Abbreviations: RTKs, receptor tyrosine kinases; EGF, epidermal growth factor; EGFR, EGF receptor; HB-EGF, heparin-binding EGF-like growth factor; TGF-α, transforming growth factor-α; AR, amphiregulin; BTC, beta-cellulin; EPR, epiregulin, NRG, neuregulin; DCM, dilated cardiomyopathy; AV, atrio-ventricular; OFT, outflow tract; EMT, endothelial-mesenchymal transformation; BMP, bone morphogenetic protein; FGF, fibroblast growth factor; HSPGs, heparan sulfate proteoglycans; HA, hyaluronic acid. ‘NRG’ refers to NRG-1 unless explicitly indicated. ‘EGF family’ refers to all ligands for ErbB receptors, and ‘EGFR-ligands’ refers to the ligands that can bind to EGFR, to distinguish from NRG that bind to ErbB3 and ErbB4, but not to EGFR. |
A fundamental requirement of biological systems is the ability to translate cues from the extracellular environment into cellular responses. Receptor tyrosine kinases (RTKs) mediate the transduction of many of these signals and are involved in such diverse processes as cell proliferation, differentiation, migration, survival, and death. RTKs are single-pass transmembrane proteins with an extracellular ligand-binding domain and an intracellular tyrosine kinase domain. Ligand binding induces homo- or heterodimerization of the receptor, which is essential for tyrosine kinase activity. The recruitment of target proteins to the activated receptor complex then initiates a signaling cascade that regulates downstream transcriptional programs.
The ErbB family of RTKs consists of four receptors: EGFR/ErbB1/HER1, ErbB2/HER2/neu, ErbB3/HER3, and ErbB4/HER4. The EGF family of ligands binds and activates ErbB receptors, resulting in the formation of homo- and heterodimers and autophosphorylation of specific tyrosine residues within the cytoplasmic domain (for a review, see Riese and Stern, 1998). Phosphorylation of these tyrosine residues allows for the binding of adapter proteins, which mediate the downstream signaling pathways that elicit a variety of biological responses. Multiple EGF family members can act as ErbB receptor ligands. Besides EGF itself, transforming growth factor-α (TGF-α), amphiregulin (AR), heparin-binding EGF-like growth factor (HB-EGF), betacellulin (BTC), epiregulin (EPR), and neuregulins (NRG1-4) can bind ErbB RTKs. EGF family members share a conserved receptor-binding motif, and soluble forms are usually produced from bioactive, integral membrane precursors (for a review see Massague and Pandiella, 1993).
In vertebrates, the different EGF family members bind to the various ErbB receptors with differing degrees of preference (Fig. 1A). For example, EGF, TGF-α, and AR bind to EGFR, whereas HB-EGF, EPR, and BTC bind to both EGFR and ErbB4. NRG-1 and NRG-2 bind to ErbB3 and ErbB4, while NRG-3 and NRG-4 bind to ErbB4, but not to ErbB3. EGF family members vary in their ability to activate distinct ErbB heterodimers, which may partly account for differences in their bioactivities. Each receptor homo- or heterodimer has a different set of tyrosine autophosphorylation sites, which serve as docking sites for specific SH2-containing proteins and recruit different combinations of signaling molecules (Di Fiore et al., 1990; Olayioye et al., 2000; Yarden, 2001).
 View Details | Fig. 1. Binding specificity of members of the ErbB receptor family to members of the EGF ligand family. (A) EGF family ligands are separated into four categories by their specificity of binding to members of the ErbB receptor family. ErbB2 has no ligand. ErbB3 is deficient in kinase activity (X). ECD, extracellular domain; ICD, intracellular domain; PM, plasma membrane. (B) ErbB homo- and heterodimer combinations activated by HB-EGF. |
A further level of complexity is introduced by the existence of an ErbB receptor that can be activated only by heterodimerization with another ligand-bound family member. ErbB2 enhances and stabilizes dimerization, but does not appear to have a direct or specific ligand itself (Horan et al., 1995). Furthermore, another ErbB receptor, ErbB3, can recruit novel SH2-containing proteins, but it does not have kinase activity as a result of substitutions of critical residues in its kinase domain (Guy et al., 1994; Kim et al., 1998). ErbB2 plays a major coordinating role in this network of receptors, with each ligand-binding receptor appearing to prefer ErbB2 as its heterodimeric partner (Tzahar et al., 1996; Graus-Porta et al., 1997). This preference is further enhanced by overexpression of ErbB2, which occurs in many types of human cancer cells (Holbro et al., 2003).
Gene targeting studies in mice have shown that the ErbB receptors and their ligands are critical for several developmental and homeostatic processes (summarized in Tables I and II). Mice deficient for ErbB2, ErbB4, and NRG die at around E10.5 (embryonic day 10.5) due to defects in cardiac trabeculae formation and peripheral nervous system development (Lee et al., 1995; Gassmann et al., 1995; Meyer and Birchmeier, 1995). ErbB3 null mice have normal heart trabeculation, but have defects in valve formation (Erickson et al., 1997). Thus, ErbB3 null mice have less severe heart defects compared with ErbB2, ErbB4, or NRG null mice, and consequently survive until about E13.5 (Erickson et al., 1997; Riethmacher et al., 1997). EGFR null mice have severe defects, and die before implantation, at mid-gestation, or within 2–3 weeks of birth, depending on the genetic background (Miettinen et al., 1995; Sibilia and Wagner, 1995; Threadgill et al., 1995). Surviving EGFR null mice with a CD1 background exhibit multiple epithelial and cerebrovascular abnormalities, as well as semilunar valve enlargement (Chen et al., 2000).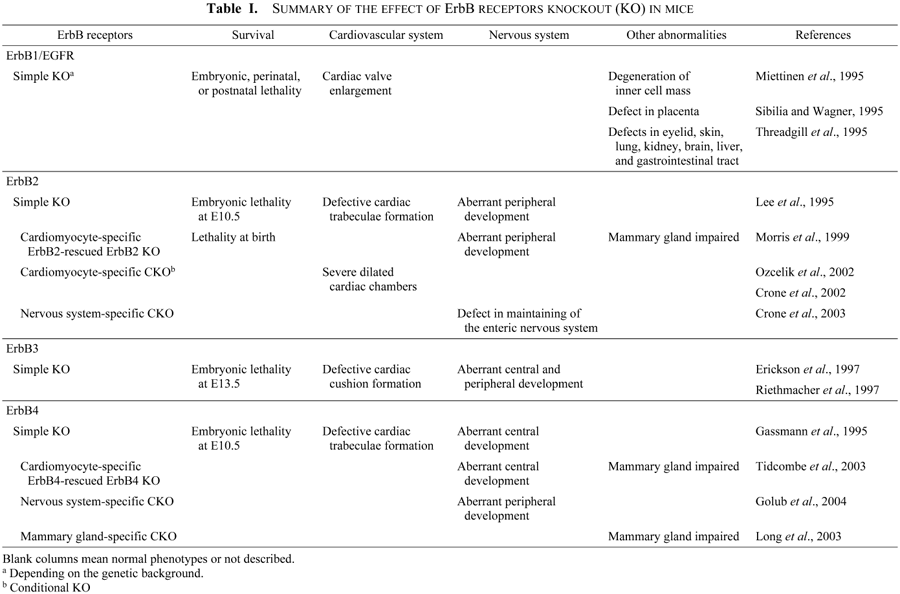
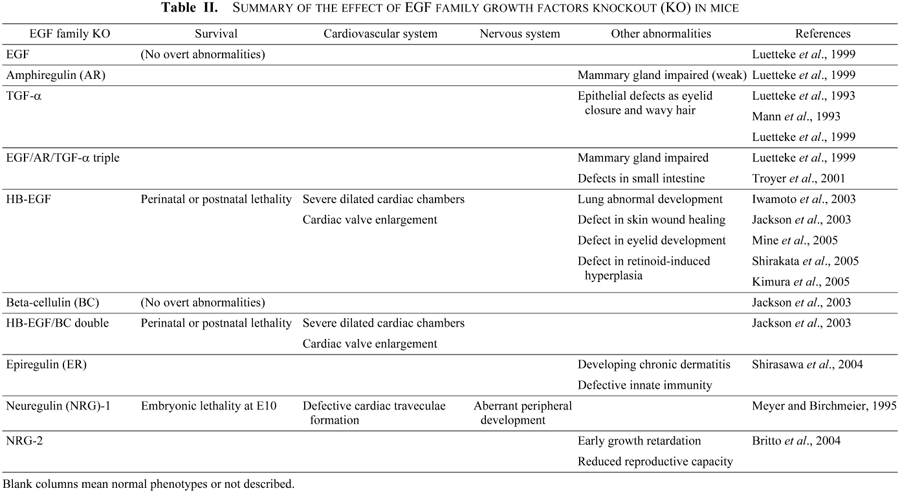
Although a number of studies have reported a critical role for ErbB receptors and NRG during development, the lethality arising from EGFR ligand deficiency during development is less well understood. For example, mice lacking either individual EGFR-ligands (EGF, TGF-α, or AR) or a combination of these ligands have revealed that: 1) TGF-α is involved in the patterning and function of hair follicles and the migration of the fetal eyelid epidermis (Luetteke et al., 1993; Mann et al., 1993); 2) AR, along with EGF and TGF-α, functions in duct morphogenesis in the developing mammary gland (Luetteke et al., 1999); 3) All three ligands are required for optimal neonatal growth, in part reflecting their role in mammary gland development, as well as their requirement for normal development and cytoprotection of the gastrointestinal tract (Troyer et al., 2001). However, even triple null mice lacking all three ligands are viable and fertile, indicating that there is considerable redundancy or cooperativity in the action of EGFR-ligands (Luetteke et al., 1999). In addition, mice lacking BTC, which is a ligand for both EGFR and ErbB4, do not show any obvious abnormalities (Jackson et al., 2003).
Deletion of EPR and HB-EGF, however, identified specific roles for these growth factors in several physiological processes. EPR null mice develop chronic dermatitis, and EPR-deficient macrophages showed a decreased response to Toll-like receptor agonists (Shirasawa et al., 2004), suggesting that EPR plays a critical role in the development and regulation of immune responses by keratinocytes and macrophages at the epidermal barrier. Meanwhile HB-EGF null mice not only exhibit abnormal lung development (Jackson et al., 2003), eyelid development (Mine et al., 2005), skin wound healing (Shirakata et al., 2005), and retinoid-induced skin hyperplasia (Kimura et al., 2005), but they also have a severe defect in heart chamber and valve formation as discussed below (Iwamoto et al., 2003; Jackson et al., 2003).
HB-EGF is a member of the EGF family of growth factors that binds to and activates EGFR and ErbB4 (Higashiyama et al., 1991; Elenius et al., 1997). HB-EGF is synthesized as a type I transmembrane protein (proHB-EGF) composed of a signal peptide, propeptide, heparin-binding, EGF-like, juxtamembrane, transmembrane, and cytoplasmic domains (Higashiyama et al., 1992) (Fig. 2). Like other EGF family members (Massague and Pandiella, 1993), the membrane-anchored form of proHB-EGF is cleaved at the juxtamembrane domain, resulting in the shedding of soluble HB-EGF (sHB-EGF) (Goishi et al., 1995). ProHB-EGF is biologically active as a juxtacrine growth factor that signals to neighboring cells in a non-diffusible manner (Higashiyama et al., 1995; Iwamoto et al., 1999; Iwamoto and Mekada, 2000). It forms complexes with CD9 and integrin α3β1 on the cell membrane (Nakamura et al., 1995), and it also functions as the receptor for diphtheria toxin (DT), mediating the entry of DT into the cytoplasm (Naglich et al., 1992; Iwamoto et al., 1994). sHB-EGF is a potent mitogen and chemoattractant for a number of cell types including vascular smooth muscle cells, fibroblasts and keratinocytes (Higashiyama et al., 1993; Raab and Klagsbrun, 1997). HB-EGF has been implicated in a number of physiological and pathological processes, which include eyelid closure (Mine et al., 2005), wound healing (Shirakata et al., 2005; Marikovsky et al., 1993; Tokumaru et al., 2000), retinoid-induced skin hyperplasia (Kimura et al., 2005), cardiac hypertrophy (Asakura et al., 2002), smooth muscle cell hyperplasia (Miyagawa et al., 1995), kidney collecting duct morphogenesis (Takemura et al., 2001), blastocyst implantation (Das et al., 1994), pulmonary hypertension (Powell et al., 1993), and oncogenic transformation (Fu et al., 1999).
 View Details | Fig. 2. The structure of HB-EGF. The mature form of HB-EGF (sHB-EGF) consists of a propeptide, a heparin-binding domain and an EGF-like domain. sHB-EGF is generated by proteolytic processing at the juxtamembrane domain (red arrow). The N-terminal propeptide is also processed (black arrow), but the biological significance of this processing is not clear. Numbers in the figure indicate amino acid residues from the N-terminus. |
While HB-EGF activates EGFR and ErbB4 directly (Higashiyama et al., 1991; Elenius et al., 1997), it can also activate ErbB2 and ErbB3 indirectly by heterodimerization (Fig. 1B). Recently, N-arginine dibasic convertase, a cell surface associated enzyme, was identified as a specific HB-EGF receptor that enhances HB-EGF-induced cell migration via EGFR (Nishi et al., 2001). Interestingly, HB-EGF also plays a critical role in the transactivation of EGFR by G protein coupled receptor (GPCR) ligands, such as lysophosphatidic acid (LPA), endothelin-1 (ET-1), and angiotensin-II (AngII). The ectodomain shedding of HB-EGF is required for this process (Prenzel et al., 1999; Eguchi et al., 2001), and abnormal regulation of HB-EGF shedding in adult mice appears to result in cardiovascular pathologies, including cardiac hypertrophy (Asakura et al., 2002), smooth muscle cell hyperplasia (Miyagawa et al., 1995), and pulmonary hypertension (Lemjabbar and Basbaum, 2002). Recent studies using cultured cells have revealed that ectodomain shedding of HB-EGF is regulated by several extracellular stimuli and intracellular signaling pathways (Izumi et al., 1998; Umata et al., 2001; Takenobu et al., 2003). Indeed, we demonstrated in vivo, by the generation and analyses of knock-in mice expressing transmembrane domain-truncated HB-EGF (HBΔtm), that deregulated secretion of sHB-EGF induces hyperplastic tissue abnormalities (Yamazaki et al., 2003).
Recently, we and another group independently demonstrated that HB-EGF is a critical factor for proper heart development and function through the analysis of HB-EGF null mice (Iwamoto et al., 2003; Jackson et al., 2003). In the next section, we describe and discuss the role of ErbB signaling and HB-EGF in these developmental and homeostatic processes in detail.
The heart is the first organ that forms during development and is required to support the rapidly growing embryo. A series of complex morphogenetic events in the heart promote proper hemodynamic development, and the appropriate hemodynamic environment supports heart morphogenesis. Thus, both morphogenetic events and the hemodynamic environment are necessary for the formation of a properly functioning vertebrate heart.
During gastrulation, cardiac progenitor cells derived from the mesoderm and neural crest migrate towards the anterior and anterio-lateral portion of the embryo. Specification of myocardial and endocardial (cardiac endothelial) cell precursors occurs during this stage (Fig. 3A). The cardiac cells then fuse at the ventral midline to form the linear heart tube, which consists of an incomplete outer muscular layer surrounding an inner endothelial tube (Fig. 3B). In mice, extensive remodeling of the heart tube occurs during mid-gestation. Between E8.5–E10.5, the heart tube undergoes a morphogenetic process called looping, in which the ventricular region adopts a pronounced rightward curvature (Fig. 3C). Cardiac looping positions the atrial region of the heart tube posterior to the common ventricle (Fig. 3D), setting the stage for forming an integrated four-chambered organ with separate systemic and pulmonary circuits (Fig. 3E). Maturation of the heart occurs in three consecutive developmental steps: 1) formation of the trabecular myocardial layer, 2) formation of the endocardial cushions followed by valve development and septation, and 3) growth and thickening of the outer compact ventricular chamber wall, followed by the establishment of coronary circulation. All of these processes involve reciprocal signaling between the primitive endocardial cells and the cardiomyocytes.
 View Details | Fig. 3. Schematic representation of heart development. (A) The precardiac mesoderm is specified in the right and left anterior plate mesoderm of the trilaminar germ disk after gastrulation. (B) Cardiac cells fuse at the ventral midline to form the linear heart tube. (C) The primitive heart tube generates a right-side bend (looping), and presumptive cardiac segments begin to appear segmentally. After this stage, trabeculae in the primitive ventricle are generated (indicated by red-dots), and endocardial cushions are formed (indicated by blue dots) both in the outflow tract (OFT) and atrio-ventricular canal (AVC). (D) Cardiac looping positions the atrial region of the heart tube posterior to the common ventricle, setting the stage for forming an integrated four-chambered mature heart (E). A, atrium; V, ventricle; CT, conus truncus; SV, sinus venosus; RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; ao, aorta; pa, pulmonary artery. |
Since it is beyond the scope of this brief section to discuss all the molecules and mutations that affect heart development and function (for reviews, see Brutsaert 2003; Chien and Olson, 2002), we will focus on the function of ErbB receptors and HB-EGF in heart development and function.
The trabeculae are finger-like extensions of the ventricular myocardium. The trabecular myocardium is largely responsible for the maintenance of blood flow during the early stages of cardiac morphogenesis before the expansion of the compact zone. Trabeculation of the spongy tissue of the heart constitutes the first step in cardiac maturation, and endocardial-myocardial signaling is necessary for this to occur. ErbB2-, ErbB4-, and NRG-null mice die at around E10.5 due to aberrant cardiac and peripheral nervous system development (Lee et al., 1995; Gassmann et al., 1995; Meyer and Birchmeier, 1995). The trabeculae fail to develop in these mice and, as a result, the heart develops an arrhythmia, an enlarged common ventricle, and reduced blood flow. Expression of ErbB2 and ErbB4 under the control of cardiac-specific promoters in ErbB2 and ErbB4 mutants, respectively, is able to rescue the cardiac defects of the mutants. ErbB2 null mice expressing ErbB2 under the cardiac specific myosin heavy chain (MHC) promoter do not show embryonic lethality, although they undergo a severe loss of both motor and sensory neurons and die at birth (Morris et al., 1999). On the other hand, rescued ErbB4 null mice are viable, but display abnormalities in the central nervous system and mammary glands (Tidcombe et al., 2003). Taken together, these indicate that signaling by NRG, ErbB2, and ErbB4 is essential for trabeculae formation.
While ErbB2 and ErbB4 function in trabeculation, ErbB3 and EGFR are important for cardiac valve formation. ErbB3 null embryos die at E13.5 due to defective cardiac cushion formation (Erickson et al., 1997), while EGFR null embryos have enlarged cardiac valves (Chen et al., 2000; Jackson et al., 2003), possibly due to deregulation of the valve remodeling process (Jackson et al., 2003). Thus, ErbB3 and EGFR seem to function sequentially in cushion formation and valve remodeling. ErbB function in cardiac valve formation is discussed in greater detail in section 3.
As in the developing heart, various factors and signaling pathways function to maintain homeostasis in the mature heart. Investigation of human cardiac diseases has provided clues as to the mechanism governing this homeostatic process. One of the most fatal cardiac diseases is dilated cardiomyopathy (DCM), which is characterized by dilated ventricles, reduced systolic function, and impaired contraction (Seidman and Seidman, 2001; Towbin and Bowles, 2002). A variety of factors, including genetic, immunologic, viral, and environmental, can cause DCM. Approximately 30% of idiopathic DCM is inherited, and more than 15 loci for human DCM have been identified. Among these are genes encoding sarcomeric cytoskeletal proteins; however, mutations in these genes account for only a minor proportion of heritable cardiomyopathies in humans (Seidman and Seidman, 2001). Recent studies in mice indicated that mutations in several non-cytoskeletal factors, such as bradykinin-B2 and serotonin-2B receptor genes, also cause DCM (Emanueli et al., 1999; Nebigil et al., 2001).
ErbB2 was recently shown to be required for the maintenance of normal heart function in adults. Since ErbB2 null embryos die during mid-gestation, a conditional gene targeting approach has been used to specifically inactivate ErbB2 in the ventricle. These mice have severe heart failure with dilated ventricles and decreased contractility (Crone et al., 2002; Ozcelik et al., 2002), symptoms resembling human DCM. In addition to ErbB2 null mice, conditional ErbB4 null mice also develop a DCM-like phenotype (‘personal communication’ in Ozcelik et al., 2002), suggesting that ErbB2 may partner with ErbB4 to form a heterodimer in the adult heart. These data support a model in which signaling through ErbB2 as well as ErbB4 is crucial for adult heart function. The monoclonal anti-ErbB2 antibody trastuzumab has recently begun being used in the treatment of breast cancer (Slamon et al., 2001). In light of the cardiac side effects associated with this treatment, an increased understanding of the mechanisms by which ErbB2 regulates heart function is eagerly awaited.
Interestingly, HB-EGF acts as the upstream factor for ErbB receptors in the adult heart. The hearts from HB-EGF null (HBdel/del) mice are grossly enlarged (Iwamoto et al., 2003) (Fig. 4A), with adult mutants demonstrating progressive ventricular dilation of both chambers (Fig. 4B). In addition, the size of cardiomyocytes was enlarged by about two-fold in HBdel/del mice compared to control mice. Cardiovascular function in these mice, as evaluated by transthoracic echocardiography (Iwamoto et al., 2003), was impaired, with the ventricular fractional shortening (FS), a measure of systolic function, was reduced to approximately half that of WT hearts. Taken together, the histopathological and echocardiographic analysis indicated that HBdel/del mice exhibit increased chamber size, myofiber hypertrophy, and decreased fractional shortening, signs of severe cardiac dysfunction. Overall, these mice had symptoms resembling the cardiac phenotype of ErbB2 conditional knockout mice.
 View Details | Fig. 4. Heart defects in HB-EGF null mice. (A) Representative adult (12-week-old) wild-type (+/+) and HB-EGF null (del/del) hearts. A surviving HB-EGF null mouse shows massive enlargement of the heart. (B) Hematoxylin/eosin staining of transverse sections of hearts from wild-type (+/+) and HB-EGF null (del/del) mice. The HB-EGF null mouse exhibits progressive dilation of both ventricular chambers. |
The phenotype of ErbB2 conditional knockout mice is typically attributed to a loss of NRG activity since NRG is a ligand for heterodimers containing ErbB2. However, the loss of HB-EGF could also be responsible for this phenotype, since it is possible that HB-EGF activates ErbB2 via EGFR:ErbB2 or ErbB2:ErbB4 heterodimers in adult cardiomyocytes. Indeed, soluble HB-EGF perfused into wild-type hearts induces tyrosine phosphorylation of EGFR, ErbB2 and ErbB4 (Iwamoto et al., 2003). Furthermore, the constitutive tyrosine phosphorylation levels of ErbB2 and ErbB4 are reduced significantly in HBdel/del mice. Since cardiomyocytes strongly express HB-EGF, cardiomyocytes may provide a source of HB-EGF that maintains constitutive phosphorylation levels (Iwamoto et al., 2003).
What is the molecular mechanism underlying the cardiac dysfunction observed in the conditional ErbB2 and HB-EGF null mutants? One possibility is that HB-EGF-ErbB signaling in the heart promotes the survival of cardiomyocytes (Chien, 2000). Cardiomyocyte-specific inactivation of various genes has demonstrated that perturbation of cardiomyocyte survival pathways leads to cardiomyopathy (Hirota et al., 1999; Yussman et al., 2002). While increased apoptosis of cardiomyocytes was not observed in the hearts of conditional ErbB2 mice (Crone et al., 2002; Ozcelik et al., 2002), partial rescue of ventricular dilation and heart contractility was observed in these mice when the antiapoptotic gene Bcl-xL was overexpressed by adenoviral infection (Crone et al., 2002). However, whether this partial rescue resulted from changes in cell survival is unclear. Further experiments should be performed to determine whether impaired survival of cardiomyocytes is the primary cause of the cardiomyopathy in conditional ErbB2 and HB-EGF null mice.
Alternatively, HB-EGF-ErbB2 signaling might control the contractility/ion homeostasis of the adult heart. Components of the excitation-contraction machinery, including ion channels, are located in the T-tubule system of cardiomyocytes, where ErbB2 and ErbB4 are also found (Ozcelik et al., 2002). RTK activation is also known to influence potassium and sodium channels in other cell systems (Hilborn et al., 1998; Wischmeyer et al., 1998). Future studies could establish whether HB-EGF has ionotropic/chronotropic properties in perfused hearts. Furthermore, ErbB receptors are known regulators of many signaling cascades, including the PI3 kinase/Akt cascade, through cross-talk with the calmodulin-dependent kinase pathway as well (Feinmesser et al., 1999; Yarden and Sliwkowski, 2001). Perturbation of these pathways is known to influence cardiac contractility (Crackower et al., 2002; Zhang et al., 2002).
HB-EGF is first synthesized as a membrane-anchored form, proHB-EGF. The soluble form, sHB-EGF, is then released from the cell surface by ectodomain shedding (Goishi et al., 1995). Both sHB-EGF and proHB-EGF are thought to be biologically active, based on several in vitro studies (Higashiyama et al., 1995; Iwamoto et al., 1999; Iwamoto and Mekada, 2000). To investigate the roles of proHB-EGF and sHB-EGF in vivo, we generated mutant mice expressing an uncleavable form of proHB-EGF (HBuc) by gene targeting (Yamazaki et al., 2003). As was the case for HB-EGF null mice (Iwamoto et al., 2003), HBuc/uc mutants developed severe heart failure with dilated ventricular chambers and defects in cardiac function, indicating that cleavage of proHB-EGF into the soluble form is necessary for normal cardiomyocyte function (Yamazaki et al., 2003). Since both HB-EGF and its receptor heterodimer ErbB2/4 are expressed in cardiomyocytes, sHB-EGF secreted from cardiomyocytes may activate ErbB2/4 on the surface of cardiomyocytes in a cell-autonomous, autocrine fashion (Fig. 5). In cardiomyocytes, ADAM12 was reported to act as a proteinase (convertase) for proHB-EGF ectodomain shedding (Asakura et al., 2002). However, ADAM12 null mice did not show any abnormalities in heart development and function (Kurisaki et al., 2003). Thus, the physiological role for this convertase(s) in the maintenance of normal cardiomyocyte function is not clear, and further studies are necessary to clarify the mechanism of HB-EGF shedding in heart development.
 View Details | Fig. 5. Model of HB-EGF function in the myocardium. sHB-EGF is released from the plasma membrane of the cardiomyocyte by ectodomain shedding of proHB-EGF, which is mediated by an ADAM family member or other metalloprotease (green arrowhead). sHB-EGF binds to and activates ErbB2/4 heterodimers, which are enriched in the T-tubule system of cardiomyocytes, resulting in signal transduction promoting cardiomyocyte survival or contractility/ion homeostasis. |
Cardiac valve formation occurs in two consecutive steps: cardiac cushion formation and valve remodeling (for reviews, see Schroeder et al., 2003; Armstrong and Bischoff, 2004). Following cardiac looping (see section 2-1), the cardiac cushions are formed from localized expansions of the extracellular matrix (cardiac jelly) in the region between the common atrium and ventricle as well as in the distal portion of the outflow tract (OFT). The subsequent transformation of a subpopulation of endocardial cells that line the cushions is essential for the development of valve leaflets and efficient valve performance. An endocardial-to-mesenchymal transformation (EMT) occurs at the atrioventricular (AV) boundary to initiate formation of the mitral and tricuspid valves, and somewhat later in the OFT to form the aortic and pulmonary valves. This process depends on soluble factors derived from the myocardium that diffuse through the cardiac jelly to signal to the endocardium. Having received signals from the myocardium, the endocardium undergoes EMT, and begins secreting soluble factors to promote the further differentiation of the cardiac cushions (Fig. 7). Several complex signaling pathways have been implicated in the EMT, including TGF-β signaling (Nakajima et al., 2000), Wnt/β-catenin signaling (Hurlstone et al., 2003), Notch signaling (Timmerman et al., 2004), NFAT and VEGF signaling (Chang et al., 2004), and ErbB signaling (Erickson et al., 1997) (as described in section 3-2). Following migration into the cardiac jelly, the mesenchymal cells form multiple layers, resulting in the expansion of the cushion crests towards one other. These cells then proliferate to form the cushions that subsequently give rise to the cardiac valves as well as the septa of the four-chambered heart (Eisenberg and Markwald, 1995; Lamers et al., 1995).
In mice, endocardial cushion formation is complete by E12.5 (Lakkis and Epstein, 1998), and it is followed by remodeling of the cushions to thin valve leaflets. Transformed AV canal cushion cells invade the extracellular matrix to initiate reshaping of the primitive tissue into valve leaflets, eventually comprising a substantial portion of the mature valves. The presumptive valves are believed to form by delaminating from the ventricular myocardial wall (Wenink and Gittenberger-de Groot, 1986). Thin strands of elongated muscle remain tethered to the valve tissue with thickened trabecular sides on the ventricular myocardial wall. These structures become the chordae and papillary muscle, respectively, of the mature valve. While much has been learned about the signaling pathways involved in the process of cushion formation, the molecular mechanisms underlying valve remodeling are poorly understood.
Several lines of evidence indicate that ErbB receptor signaling is required during cardiac cushion formation, in addition to myocyte maturation. Expression of NRG is restricted to the cardiac endocardium (Meyer and Birchmeier, 1995), and ErbB2 is expressed in embryonic heart tissues (Erickson et al., 1997; Camenisch et al., 2002). EGFR has a global expression pattern in embryonic heart valve tissues (Jackson et al., 2003), whereas ErbB3 appears to be restricted to the endocardium and transformed mesenchymal cells during AV canal mesenchyme production (Erickson et al., 1997), and ErbB4 is expressed in the myocardium (Gassmann et al., 1995). These different and restricted expression patterns during heart morphogenesis suggest that these molecules have distinct functions during muscle and valve formation. Gene targeting studies revealed that embryos lacking ErbB3 have cardiovascular defects, including AV canal malformation (Erickson et al., 1997). These cardiac cushion abnormalities result in blood reflux through the defective valve tissue, contributing to the lethality of ErbB3 null embryos by E13.5. In contrast, ErbB4 null embryos do not have valve defects, suggesting that it is not essential for valvulogenesis (Gassmann et al., 1995). Although not as pronounced as the ErbB3 mutant cushion phenotype, embryos deficient for ErbB2 or NRG also exhibit underdeveloped cushion and valve tissue, but die at E10.5, presumably due to failed myocyte differentiation and contractile deficiencies (Erickson et al., 1997). These data suggest that ErbB2/3 receptor heterodimers function in the early stages of cardiac valve formation.
In addition, extracellular matrix components in the cardiac jelly modulate the ErbB signaling that regulates cushion formation. Hyaluronic acid (HA), a major component of cardiac jelly, has been implicated in ErbB3 signaling during cushion formation (Camenisch et al., 2002). HA is a glycosaminoglycan composed of alternating glucuronic acid and N-acetylglucosamine residues, and Has2 appears to be the major enzyme responsible for HA synthesis during development. Has2 null mice die by E9.5 and display pericardial edema, disordered vessel growth, and the complete absence of cardiac jelly (Camenisch et al., 2000). NRG was able to rescue the Has2 null phenotype in cushion explant experiments (Camenisch et al., 2002). Furthermore, Has2 null mice show decreased ErbB2 and ErbB3 phosphorylation in endocardial cushions relative to wild-type embryos. Addition of HA to Has2 null tissue explants was able to restore ErbB3 phosphorylation (Camenisch et al., 2002). Together, these findings suggest that HA in cardiac jelly regulates ErbB2/3 signaling activity, which may be triggered by NRG signals from the endocardium, in particular the mesenchymal cells undergoing EMT (Fig. 7).
EGFR signaling appears to function at a later stage during valve formation, the valve remodeling process. Normal semilunar (aortic and pulmonic) valve development requires EGFR tyrosine kinase signaling (Chen et al., 2000), and recent observations of EGFR deficient mice show more global abnormalities in both AV (mitral and tricuspid) and semilunar valves in these mutants (Jackson et al., 2003). A similar requirement for EGFR in valve formation has also been observed in zebrafish. Addition of EGFR kinase inhibitors or the transient knockdown of EGFR expression with morpholino resulted in decreased circulation by 80 hours post fertilization. Microangiography revealed a narrow OFT with massive ventricular and atrial enlargement, likely secondary results of increased afterload (Goishi et al., 2003). It is not yet clear whether the narrowed OFT results from thickened OFT cushions or an alteration of neural crest cell migration. However, it appears that the cardiac-specific phenotype of EGFR disruption is conserved across multiple experimental models.
Consistent with the valve defect in EGFR null animals, HB-EGF null mice showed cardiac valve abnormalities (Iwamoto et al., 2003; Jackson et al., 2003). HB-EGF is expressed exclusively in endocardial cells, but not in differentiated mesenchymal cells, during embryonic heart valve development (Iwamoto et al., 2003) (Fig. 6). Histological analysis of embryonic HB-EGF null hearts showed enlarged semilunar valves and AV valves with an abnormal thickened morphology (Iwamoto et al., 2003; Jackson et al., 2003) (Fig. 6). Masson-trichrome staining indicated that no particular fibrosis occurred in the enlarged valves (Iwamoto et al., 2003), suggesting that valve thickening resulted from an increased number of mesenchymal cells.
 View Details | Fig. 6. Cardiac valve enlargements in HB-EGF null mice. Longitudinal sections through cardiac valves of E17.5 LacZ-stained heterozygote (del/+) and homozygote (del/del) embryos are shown. The targeting vector used to delete HB-EGF contained the lacZ gene, which was used as a reporter for HB-EGF expression. LacZ reporter expression is observed exclusively in the endocardium. HB-EGF null hearts show enlarged semilunar (aortic) valves and AV (tricuspid) valves with an increased number of cushion mesenchymal cells. Pulmonic and mitral valves are also enlarged (not shown). |
HB-EGF is thought to function in the valve remodeling process rather than in cushion formation (Jackson et al., 2003). Analysis of HB-EGF null mice suggests that HB-EGF might regulate the extent of mesenchymal cell proliferation during valve remodeling. Evidence for this includes the following: 1) EMT in cushion formation occurs normally in HB-EGF null mice, 2) the rate of apoptosis is unchanged in HB-EGF null valves during remodeling, and 3) excessive proliferation of the mesenchymal cells occurred in HB-EGF null valves (Jackson et al., 2003).
Aberrant hyperproliferation of transformed mesenchymal cells in HB-EGF- and EGFR-deficient cardiac valves suggests that HB-EGF signaling through EGFR negatively regulates proliferation of the mesenchymal cells during valve remodeling. What is the mechanism underlying HB-EGF-EGFR signaling in valve remodeling? It has been reported that cardiac valve formation depends on signaling by the bone morphogenetic protein (BMP) family of secreted signaling molecules, which are members of the TGFβ superfamily. Several lines of evidence from BMP mutant mice suggest that valves fail to form without sufficient BMP signaling, but valves are hyperplastic when BMP signaling is deregulated (Clark et al., 1999; Delot et al., 2003; Kim et al., 2001; Galvin et al., 2000). Moreover, the ability of EGFR to regulate BMP signaling has been reported in cell culture studies (Kretzschmer et al., 1997; Nonaka et al., 1999; Lo et al., 2001). Interestingly, increased activation of nuclear Smad1, 5, and 8, the intracellular mediators of BMP signaling, was observed in the hyperplastic valves of HB-EGF mutants (Jackson et al., 2003). Further studies are necessary to clarify the relationship between BMP signaling and HB-EGF-EGFR signaling during cardiac valve formation.
Another candidate for the target of HB-EGF-EGFR signaling in valve leaflet remodeling is fibroblast growth factor (FGF)-4 (Sugi et al., 2003). In the chick embryo, FGF-4 protein was detectable in the cushion mesenchymal cells as well as in the myocardial cells, while FGF receptor (FGFR) 2 is expressed exclusively in cushion mesenchymal cells (Sugi et al., 2003). Moreover, it was demonstrated both in vitro and in vivo that FGF-4 can induce proliferation of mesenchymal cells during chick early valve leaflet formation (Sugi et al., 2003). Although the relationship between FGF-FGFR signaling and HB-EGF-EGFR signaling remains unclear, it is worth investigating whether there is cross-talk between these two signaling cascades in mouse valve formation.
Does EGFR partner with another ErbB receptor to regulate valve formation? The lack of valve defects in ErbB2 or ErbB4 null mice rescued by cardiomyocyte-specific expression of ErbB2 or ErbB4 suggests that these receptors are not essential for valve formation (Morris et al., 1999; Tidcombe et al., 2003). The underdeveloped cushions of ErbB3 null hearts, in contrast, indicate that this receptor is required for an early step in valve formation (Erickson et al., 1997; Camenisch et al., 2002). This does not exclude a possible requirement for ErbB3, possibly in partnership with EGFR, during this stage of valve remodeling. Future studies should be aimed at determining whether EGFR functions as a homo- or heterodimer in this context.
During cardiac valve development, HB-EGF appears to act as a negative regulator for the proliferation of mesenchymal cells. The membrane-anchored form of HB-EGF (proHB-EGF) is known to inhibit the proliferation of certain types of EGFR-expressing cells in a juxtacrine fashion (Iwamoto et al., 1999). Thus, one can expect that the process of valve formation might be mediated by proHB-EGF. However, this was not the case. As was the case for maintenance of mature heart function, it was shown by mutant mice expressing the uncleavable form of proHB-EGF (HBuc/uc) that generation of the soluble form of HB-EGF is essential for valve formation (Yamazaki et al., 2003). The requirement of the soluble form of HB-EGF in this process was further supported by the analysis of mice lacking ADAM17/TACE. ADAM17 is a putative proHB-EGF convertase, and mice lacking ADAM17 showed enlarged valves quite similar to those of HB-EGF null mice (Jackson et al., 2003; Sahin et al., 2004). Although it remains unclear whether the valve formation defects in ADAM17 null mice result from a failure of ADAM17 to directly cleave proHB-EGF, these findings strongly suggest that ADAM17 is a key enzyme for the ectodomain shedding of proHB-EGF in valve formation (Jackson et al., 2003; Sahin et al., 2004), and that HB-EGF might act as a paracrine factor, not a juxtacrine factor, in valve formation (Yamazaki et al., 2003).
HB-EGF is expressed specifically in endocardial cells, but not in differentiated mesenchymal cells (Iwamoto et al., 2003). Conversely, EGFR is expressed in mesenchymal cells as well as in endocardial cells (Jackson et al., 2003). No abnormalities in endocardial cells were observed in HB-EGF null valves, suggesting that sHB-EGF secreted from endocardial cells diffuses into the cardiac jelly and acts upon EGFR-expressing differentiated mesenchymal cells during valve remodeling (Fig. 7). Notably, HB-EGF has heparin-binding properties, and the association of heparan sulfate proteoglycans (HSPGs) with HB-EGF modulate the biological activity of this growth factor (Higashiyama et al., 1993; Raab and Klagsbrun, 1997; Aviezer and Yayon, 1994; Shishido et al., 1995; Takazaki et al., 2004). Cardiac jelly is a rich in extracellular matrix components, including HSPGs as well as HA (Meier et al., 1998), suggesting the possibility that HSPGs in cardiac jelly are also involved in cardiac valve formation in association with HB-EGF.
 View Details | Fig. 7. Possible model of ErbB and HB-EGF function in cardiac valve formation. During cardiac cushion formation, the transformation of a subpopulation of endocardial cells is initiated by several soluble factors derived from the myocardium (a). Subsequent EMT and migration of transformed mesenchymal cells toward the cardiac jelly is mediated by ErbB2/3-signaling (b). sNRG is released from the plasma membrane of endocardial cells by ectodomain shedding of proNRG, which is mediated by an unknown protease (pink arrowhead). sNRG then binds to and activates ErbB2/3 heterodimers on mesenchymal cells. This process is mediated by hyaluronan in the cardiac jelly, resulting in signal transduction that promotes transformation and migration. After EMT, HB-EGF-EGFR signaling is required for appropriate valve remodeling (c). sHB-EGF is released from the plasma membrane of the endocardium by ectodomain shedding of proHB-EGF, which is mediated by ADAM17 (pink arrowhead). sHB-EGF binds to and activates EGFR on mesenchymal cells, resulting in signal transduction that suppresses mesenchymal cell proliferation. |
A number of studies as described in this review indicate that EGF-ErbB signaling is critical for normal heart development and function. During mid-gestation, NRG-ErbB2/4 signaling functions in cardiac trabeculae formation, and NRG-ErbB2/3 signaling functions in cardiac cushion formation. On the other hand, HB-EGF-EGFR signaling functions in cardiac valve remodeling after cushion formation. During and after the perinatal stages, HB-EGF-ErbB2/4 signaling functions in the maintenance of cardiomyocytes. It is likely that there is cooperation between ErbB signaling and several other signaling cascades, and cross-talk likely occurs between the signaling cascades downstream of ErbBs.
ErbB receptors are widely viewed as possible effective targets for cancer therapy. Although it was not mentioned in this review, among all of the EGFR-ligands, HB-EGF plays a key role in the progression of ovarian cancer (Miyamoto et al., 2004). Thus, HB-EGF is a novel target for the therapy of ovarian cancer. Indeed, a specific anti-HB-EGF reagent CRM197, a non-toxic mutant of diphtheria toxin, completely blocks tumor formation of ovarian cancer cells (Miyamoto et al., 2004). However, as mentioned in this review, among EGF family growth factors, HB-EGF is a critical factor for the maintenance of heart function. Thus, cardiac side effects must be strongly considered when anti-HB-EGF reagents are used for anti-cancer therapy, as was the case with trastuzumab, an anti-ErbB2 reagent. On the other hand, the requirement for the release of soluble HB-EGF for normal heart function and the DCM-like phenotype in HB-EGF null mice suggest that HB-EGF, its processing enzyme, and signaling molecules downstream of HB-EGF could be potential therapeutic targets for patients with DCM. In the future, HB-EGF and the careful regulation of its activity will be strong therapeutic targets for several distinct disease pathologies.
|