| To whom correspondence should be addressed: Eisuke Mekada, Department of Cell Biology, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan. Tel: +81–6–6879–8286, Fax: +81–6–6879–8289 E-mail: emekada@biken.osaka-u.ac.jp Abbreviations: EGF, epidermal growth factor; HB-EGF, heparin-binding EGF-like growth factor; TPA, 12-O-tetradecanoylphorbol-13-acetate; EGFR, EGF receptor; N, amino; C, carboxyl; DT, diphtheria toxin; LPA, lysophosphatidic acid; HA, hemagglutinin; PKC, protein kinase C; TGF-α, transforming growth factor-α; AR, amphiregulin; BTC, β-cellulin; ER, epiregulin; PLZF, promyelocytic leukemia zinc finger protein. |
Heparin-binding EGF-like growth factor (HB-EGF) is a member of the EGF family of growth factors that acts as a potent mitogen and chemotactic factor for fibroblasts and smooth muscle cells (Higashiyama et al., 1991; 1993). It has also been shown to play essential roles in heart and lung development and function (Iwamoto et al., 2003; Jackson et al., 2003), skin wound healing (Shirakata et al., 2005) and hyperplasia (Kimura et al., 2005), and eyelid formation in mice (Mine et al., 2005). In addition, aberrant expression of HB-EGF leads to malignant growth of carcinoma cells (Ongusaha et al., 2004; Miyamoto et al., 2004; Yagi et al., 2005; Tanaka et al., 2005).
HB-EGF is first synthesized as a transmembrane precursor protein whose extracellular portion is composed of a prodomain followed by heparin-binding, EGF-like, and juxtamembrane domains. The membrane-anchored form of HB-EGF (proHB-EGF) acts as a diphtheria toxin (DT) receptor (Naglich et al., 1992); DT bound to proHB-EGF undergoes endocytosis and inhibits protein synthesis through ADP-ribosylation of elongation factor-2. However, consistent with its paracrine and autocrine functions, soluble HB-EGF can also be produced by protease-mediated cleavage within the extracellular juxtamembrane domain. This process, termed ectodomain shedding, can be stimulated by various physiological and pharmacological agonists, including 12-O-tetradecanoylphorbol-13-acetate (TPA) (Goishi et al., 1995), calcium ionophore (Dethlefsen et al., 1998), and G protein coupled receptor ligands such as lysophosphatidic acid (LPA) (Prenzel et al., 1999; Umata et al., 2001). Cellular stresses caused by interleukin-1β, reactive oxygen, and osmotic shock also induce ectodomain shedding of HB-EGF (Takenobu et al., 2003; Fischer et al., 2004). Recent studies demonstrated that ectodomain shedding of HB-EGF is necessary for normal development in mice and tumorigenic activity of HB-EGF (Miyamoto et al., 2004; Yamazaki et al., 2003).
Although the physiological and pathological functions of soluble HB-EGF and the mechanisms of ectodomain shedding have been extensively studied, the biological role of proHB-EGF and the transmembrane and cytoplasmic domains are still largely unknown. Deletion of the transmembrane and cytoplasmic domains of proHB-EGF leads to abnormal development in mice (Yamazaki et al., 2003), indicating that the transmembrane domain is necessary for the regulation of soluble HB-EGF levels by tethering proHB-EGF on the plasma membrane. In addition, it has been suggested that proHB-EGF contributes to inhibition of cell growth (Miyamoto et al., 2004; Iwamoto et al., 1999) or mediates cell-cell adhesion (Singh et al., 2004).
Several lines of evidence have suggested roles for the cytoplasmic domain of HB-EGF and other EGF receptor (EGFR) family ligands, which include transforming growth factor-α (TGF-α), amphiregulin (AR), and neuregulins. It has been reported that the cytoplasmic domain of TGF-α binds to syntenin/mda-9/TACIP18 (proTGF-α cytoplasmic domain-interacting protein 18) (Fernandez-Larrea et al., 1999), GRASP55 (Golgi reassembly stacking protein of 55 kDa) (Kuo et al., 2000), and MAGI-3 (membrane associated guanylate kinase inverted-3) (Franklin et al., 2005), and that these binding proteins may be involved in maturation and transport of membrane proteins to the cell surface. The cytoplasmic domain of AR appears to be required for basolateral sorting, but not for constitutive and induced ectodomain shedding (Brown et al., 2001). Furthermore, it has been reported that dimerization via the cytoplasmic domain is necessary for proper ectodomain shedding of neuregulins (Liu et al., 1998). The cytoplasmic domains of neuregulins can also bind to LIM kinase, although their function remains unknown (Wang et al., 1998). In the case of HB-EGF, a multi-functional protein BAG-1 has been reported to bind to the cytoplasmic domain of HB-EGF and increase secretion of HB-EGF (Lin et al., 2001). Recently, it has been suggested that the C-terminal fragment of HB-EGF generated by ectodomain shedding translocates to the nucleus and promotes S-phase entry by sequestering promyelocytic leukemia zinc finger protein (PLZF), a transcriptional repressor of cyclin A (Nanba et al., 2003).
In this study, we demonstrate that a serine residue within the cytoplasmic domain of HB-EGF is phosphorylated by stimuli that induce ectodomain shedding, and that a mutation of the phosphorylation site reduces HB-EGF-dependent tumorigenesis in nude mice.
12-O-tetradecanoylphorbol-13-acetate (TPA), hygromycin B, and EZview red anti-HA affinity gel were purchased from Sigma (St. Louis, MO, USA). GM6001, orthovanadate and phenylarsine oxide were from Calbiochem (San Diego, CA, USA). NaF, protease inhibitor cocktail, and puromycin were purchased from Nacalai Tesque (Kyoto, Japan). Phosphorus-32 and Iodine-125 were purchased from Amersham Biosciences (Buckinghamshire, UK). Goat polyclonal antibody against the C-terminus of human pro-HB-EGF (C-18) and rabbit anti-HA polyclonal antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat anti-human HB-EGF neutralizing polyclonal and mouse anti-human HB-EGF monoclonal antibodies were obtained from R&D Systems (Minneapolis, MN, USA). Mouse anti-actin monoclonal antibody was purchased from Chemicon (Temecula, CA, USA). Anti-ERK and anti-phosphoERK antibodies were obtained from Cell Signaling (Beverly, MA, USA). Sulfo-NHS biotin was obtained from Pierce (Rockford, IL, USA). Blasticidin S was from InvivoGen (San Diego, CA, USA).
Retrovirus vectors pCX4pur (Akagi et al., 2002), pCX4bsr/SV40 T, and ecotropic retrovirus receptor cDNA in pCX4hyg were provided by T. Akagi (Osaka Bioscience Institute, Osaka Japan). Hemagglutinin (HA)-tagged HB-EGF cDNA was constructed as follows: oligonucleotide coding HA epitope sequence was ligated into XbaI-NotI sites of pBluescript-SK (+) (Stratagene, La Jolla, CA, USA). Human HB-EGF cDNA was amplified to remove stop codon and cloned into EcoRI-XbaI sites of the HA-tagging vector, and HA-tagged HB-EGF cDNA was subcloned into EcoRI-NotI sites of pCX4pur vector. Mutants with Ala substitutions at Thr205, Ser207, and Ser209 were generated by PCR-based site-directed mutagenesis. TGF-α and amphiregulin cDNA were gifts from M. Klagsbrun (Children’s Hospital, Harvard Medical School, Boston, MA, USA). Epiregulin cDNA was a gift from T. Sasazuki (Kyushu University, Fukuoka, Japan). β-cellulin cDNA was cloned from A431 cells by RT-PCR. TGF-α and amphiregulin were tagged with HA after PCR using the tagging vector and subcloned into BamHI-NotI sites of pCX4pur. Epiregulin and β-cellulin were tagged with HA after PCR using the tagging vector and subcloned into EcoRI-NotI sites of pCX4pur.
Vero cells were maintained with modified Eagle’s medium (MEM) supplemented with nonessential amino acids, 10% fetal calf serum (FCS), streptomycin, and penicillin. Buffalo rat liver (BRL) cells (provided by K. Miyazaki, Yokohama City University, Yokohama, Japan) and retrovirus packaging cell line Plat-E (Morita et al., 2000) were maintained with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FCS, streptomycin, and penicillin. To generate Vero cells susceptible to ecotropic retrovirus infection, Vero cells were transfected with ecotropic retrovirus receptor cDNA by the calcium phosphate method and selected with medium containing hygromycin B. Mouse embryonic fibroblasts (MEFs) were obtained from E13.5 HB-EGFdel/del embryos (Iwamoto et al., 2003). After 4–5 passages, the MEFs were immortalized by infection with retrovirus encoding SV40 T-antigen, and selected with DMEM supplemented with 10% FCS, streptomycin, penicillin, and blasticidin S. A selected clone was used in this study. Vero cells expressing ecotropic retrovirus receptor, BRL, and MEF cells were infected with control, or wild-type or mutant HB-EGF as described below. After infection, cells were selected with medium containing puromycin (2–10 μg/ml) for a week. Mixed, nonclonal populations of cells were used in all experiments unless otherwise noted.
Plat-E was transfected with retrovirus vectors using FuGene6 (Roche Diagnostics, Tokyo, Japan) according to the manufacturer’s directions. Two days after transfection, culture supernatants were collected, filtered, and stored at –30°C until use. Cells were infected with retroviruses in the presence of 8 μg/ml of polybrene. Two days after infection, cells were selected in media containing appropriate antibiotics.
Vero cells expressing HA-tagged HB-EGF treated with TPA were lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 20 μg/ml antipain, 10 μg/ml chymostatin, and protease inhibitor cocktail) on ice. Phosphatase inhibitors (50 mM sodium diphosphate, 10 mM NaF, 2 mM orthovanadate, and 0.5 mM PAO) were added to the lysis buffer when indicated. Cell lysates were clarified by centrifugation at 39,000×g and loaded to 0.5 ml of EZview red anti-HA affinity gel. Columns were washed by sequential two steps with the following buffers (20 bed volumes/step): 10 mM Tris-HCl, pH 7.4, 0.2 M NaCl, 0.1% SDS; and 10 mM Tris-HCl, pH 7.4, 3 M NaCl. Bound material was eluted 5 times with 1 bed volume of 0.2 M glycine, pH 2.5, and neutralized with Tris-HCl, pH 8.8. N-terminal amino acid sequences of C-terminal fragments were analyzed with an amino acid sequencer (model 492cLC, Applied Biosystems).
Vero cells expressing wild-type and mutant HB-EGF (5×105 cells) were seeded in a 35 mm dish, cultured for 6–8 h, and serum starved overnight. Following treatment with TPA (64 nM) for 30 min, cells were lysed with lysis buffer containing phosphatase inhibitors for 5 min on ice, and the lysates were clarified by centrifugation. Immunoblotting was done as described previously (Umata et al., 2001; Takenobu et al., 2003). Ectodomain shedding of HB-EGF was monitored by generation of the C-terminal fragment, using an antibody recognizing the C-terminus of HB-EGF in the case of nontagged HB-EGF, and an anti-HA antibody for HA-tagged HB-EGF. To detect soluble HB-EGF in the culture medium, soluble HB-EGF was enriched with heparin-Sepharose (Amersham Biosciences, Uppsala, Sweden) and subjected to immunoblotting analysis.
Cell surface biotinylation was performed as described previously (Nakamura et al., 1995). Cell lysates prepared as described above were incubated with mouse anti-HB-EGF monoclonal antibody and protein G Sepharose (Amersham Biosciences) for 3 h at 4°C. Immunoprecipitated samples were subjected to immunoblotting analysis with goat anti-HB-EGF neutralized antibody followed by HRP-conjugated anti-goat IgG antibody or with HRP-conjugated streptavidin.
Vero cells (5×105 cells/35-mm dish) or BRL cells (2×106 cells/6-cm dish) were cultured overnight and serum starved for 6–12 h. Cells were washed 3 times with warmed phosphate- and serum-free DMEM (Gibco, Grand Island, USA) supplemented with 25 mM HEPES, pH 7.4. Thirty minutes after incubation with phosphate- and serum-free medium at 37°C, phosphorus-32 [32P] was added to culture medium at 9.25 MBq/ml and incubated for 2 h. Cells were stimulated with 64 nM TPA in the presence of 10 μM GM6001 for 30 min when indicated. Cells were washed twice with ice-cold PBS and lysed with EDTA-free lysis buffer containing phosphatase inhibitors. Lysates were clarified by centrifugation, and supernatants were incubated with anti-human HB-EGF monoclonal antibody and protein A Sepharose or anti-HA antibody beads. During incubation, DNAase and RNAase were added to the lysis buffer to avoid contamination by phosphorylated DNA and RNA. Immunoprecipitates were washed with the lysis buffer containing phosphatase inhibitors and subjected to electrophoresis. Phosphorylation of HB-EGF and other EGFR ligands were detected by using BAS-1500 image analyzer (Fuji Photo Film Ltd., Tokyo, Japan).
DT sensitivity and binding of 125I-labeled DT to cells or heparin-Sepharose were measured as described previously (Iwamoto et al., 1994). Nonspecific binding of 125I-DT was assessed in the presence of a 3000-fold excess of unlabeled DT. Specific binding was determined by subtracting the amount of nonspecific binding from total binding obtained with 125I-DT alone. The amount of DT bound to cells was calculated from the value of the specific binding of DT.
Cells (5×106 cells) were injected into 6-week-old male BALB/cAJcl-nu/nu mice (CLEA Japan, Inc., Tokyo, Japan). Tumor weights were measured 4 or 6 weeks after injection. The handling of animals was performed in accordance with the guidelines of Osaka University.
We previously reported that TPA induces ectodomain shedding of HB-EGF in Vero cells. Amino acid composition analysis of the carboxy(C)-terminal fragment indicated the presence of a cleavage site at Pro149-Val150, in the juxtamembrane domain (Goishi et al., 1995). Here, we found that when C-terminally HA-tagged human HB-EGF was stably expressed in Vero cells, two distinct C-terminal fragments were generated upon TPA stimulation (Fig. 1A). In addition, two C-terminal fragments were generated from nontagged HB-EGF upon TPA treatment, though the slower migrating band was much less abundant than the faster migrating band. It has been reported that HB-EGF is also cleaved at Ser147-Leu148 and Leu148-Pro149 in U-937 cells (Higashiyama et al., 1992). Cell-free digestion analysis has shown that matrix metalloproteinase 3 cleaves HB-EGF at Glu151-Asn152 (Suzuki et al., 1997). Thus, we wished to test the possibility that the two C-terminal fragments were generated by alternative cleavage at two distinct sites. We purified the C-terminal fragments from TPA-treated Vero cells expressing HA-tagged HB-EGF using an anti-HA monoclonal antibody column and analyzed their N-terminal amino acid sequences. We found that the C-terminal fragments with differing mobility by SDS-PAGE both had a Val150 residue at the N-terminus. These results suggest that the two distinct types of C-terminal fragments were not generated by alternative cleavage at the juxtamembrane domain.
 View Details | Fig. 1. Generation of two C-terminal fragments of HB-EGF by TPA stimulation. (A) Ectodomain shedding of nontagged or HA-tagged HB-EGF upon TPA stimulation. Vero cells stably expressing nontagged or HA-tagged HB-EGF were treated with 64 nM TPA for 30 min. Resulting C-terminal fragments were detected by an antibody recognizing the C-terminus of HB-EGF (HB C-ter). Two C-terminal fragments were clearly detected from HA-tagged HB-EGF. In the case of nontagged HB-EGF, a faint, slower migrating band (asterisk) and a strong, faster migrating band were detected. Actin was used as a loading control. (B) Phosphatase sensitivity of the upper C-terminal fragment. C-terminal fragments were immunoaffinity purified from cell lysates of Vero cells expressing HA-tagged HB-EGF stimulated with TPA in the absence (left lane) or presence (right lane) of phosphatase inhibitors. Silver staining of purified samples show that the upper C-terminal band (asterisk) was purified only in the presence of phosphatase inhibitors. |
During the course of purification, we found that the slower migrating band could be purified only in the presence of phosphatase inhibitors (Fig. 1B). This suggests that the two bands represent different phosphorylation states. To test this idea, we first used anti-phosphotyrosine antibodies (PY20 and 4G10) to examine whether the C-terminal fragments generated by TPA stimulation underwent tyrosine phosphorylation, but were unable to detect any tyrosine phosphorylation (data not shown). We next examined whether or not serine/threonine phosphorylation might account for the two C-terminal species. There are three serine/threonine residues in the cytoplasmic domain of HA-tagged HB-EGF, Thr205, Ser207, and Ser209, with Ser209 artificially introduced by the construction of the HA-tag (Fig. 2A). We constructed three mutants of HA-tagged HB-EGF, in which Ala was substituted for Thr205, Ser207, and Ser209, and established Vero cells stably expressing these mutants. Surface biotinylation analysis indicated that cell surface expression of these mutants was not impaired by the mutations (Fig. 2B). When these cells were treated with TPA, the slower migrating band was generated from the T205A and S209A mutants, though at lower levels than wild-type (Fig. 2B). In contrast, the slower migrating band was not generated from S207A mutant, suggesting that the slower migrating fragment is phosphorylated at Ser207.
 View Details | Fig. 2. Serine phosphorylation of HB-EGF cytoplasmic domain. (A) Amino acid sequence of human HB-EGF cytoplasmic domain. The upper and lower lines indicate amino acid sequence of the nontagged and HA-tagged HB-EGF cytoplasmic domains, respectively. Amino acid residues artificially added for the construction are italicized. HA tag sequence is indicated by thick underlining. Serine and threonine are denoted by thin underlining. (B) Generation of the upper C-terminal fragment from mutants HB-EGF. Mutation at Thr205 (T205A) or Ser209 (S209A) does not abrogate generation of the upper C-terminal fragment, while mutation at Ser207 (S207A) completely impairs generation of the upper C-terminal fragment indicated with arrow (top). Actin represents loading control of the two top lanes (middle). Cell surface biotinylation shows that these HB-EGF mutants express on the plasma membrane (bottom). (C) Phosphorylation of HB-EGF. Vero cells expressing wild-type or mutant HB-EGF were incubated with medium containing [32P]-orthophosphate and then stimulated with TPA for 30 min. Cell lysates were immunoprecipitated with anti-HA antibody and immunoprecipitates were subjected to SDS-PAGE. Then radioactive bands were detected with a BAS imager. Phosphorylated band corresponding to C-terminal fragment of HB-EGF (arrow) was detected from wild-type (WT), T205A, and S209A mutants, but not from S207A mutant (top). Arrowhead indicates a nonspecific band. Immunoblotting of the samples with anti-HA antibody shows that generation of the upper fragments correlates with phosphorylation levels of the C-terminal fragment indicated by arrow (bottom). (D) Alignment of amino acid sequences of mammalian HB-EGF cytoplasmic domain. Serine residue corresponding to Ser207 of human HB-EGF is indicated with a box. |
To further examine whether Ser207 is phosphorylated upon TPA stimulation, a metabolic labeling experiment was performed using Vero cells cultured in medium containing [32P]-orthophosphate. Vero cells expressing HA-tagged wild-type or mutant HB-EGF were stimulated with TPA, and phosphorylation was monitored by autoradiography. A phosphorylated C-terminal fragment was detected from HA antibody immunoprecipitates of wild-type, T205A and S209A mutants, whereas no phosphorylated C-terminal fragment was detected from the S207A mutant (Fig. 2C). These results indicate that Ser207, which is conserved among mammals (Fig. 2D), is phosphorylated by TPA stimulation, thereby generating two distinct types of C-terminal fragments.
Since the S207A mutant is efficiently cleaved to generate the C-terminal fragment but is not phosphorylated upon TPA treatment (Figs. 2B, 2C), we concluded that phosphorylation of Ser207 is not required for TPA-induced ectodomain shedding.
To examine whether ectodomain shedding is indispensable for the serine phosphorylation, Vero cells expressing HA-tagged wild-type or S207A mutant HB-EGF were metabolically labeled with [32P]-orthophosphate, then stimulated with TPA in the presence of the metalloprotease inhibitor GM6001, which inhibits ectodomain shedding. TPA-induced ectodomain shedding of HB-EGF was almost completely inhibited by GM6001 (Fig. 3A, lower panel), and multiple radioactive bands corresponding to proHB-EGF were detected for wild-type but not S207A mutant HA-tagged HB-EGF expressing cells upon TPA treatment (Fig. 3A). A similar experiment was performed for nontagged HB-EGF. When Vero cells expressing nontagged wild-type or S207A mutant HB-EGF were treated with TPA in the presence of GM6001, ectodomain shedding was inhibited and multiple phosphorylated proHB-EGF bands were detected for wild-type but not S207A mutant HB-EGF (Fig. 3B). These results indicate that proteolytic cleavage of the juxtamembrane domain is not required for TPA-induced C-terminal serine phosphorylation. In addition, Fig. 3B further confirms that Ser207 phosphorylation occurs in nontagged proHB-EGF upon TPA treatment.
 View Details | Fig. 3. Extracellular stimuli inducing ectodomain shedding of HB-EGF causes C-terminal serine phosphorylation. (A) Effect of metalloprotease inhibitor on serine phosphorylation. Vero cells expressing HA-tagged wild-type or S207A mutant HB-EGF were incubated with [32P]-orthophosphate and stimulated with TPA in the presence of metalloprotease inhibitor GM6001 (10 μM). HB-EGF was immunoprecipitated with anti-HA antibody and analyzed by SDS-PAGE followed by detection with a BAS imager. Multiple phosphorylated bands corresponding to proHB-EGF were detected from the wild-type but not the S207A mutant form (top). Immunoblotting of the same samples with anti-HA antibody confirmed that TPA did not affect total HB-EGF levels (bottom). (B) Phosphorylation of nontagged HB-EGF. Vero cells expressing nontagged wild-type HB-EGF or S207A mutant HB-EGF were incubated with [32P]-orthophosphate and stimulated with TPA as described above. Cell lysates were immunoprecipitated with control antibody (–) or an anti-HB-EGF antibody recognizing the ectodomain of HB-EGF (+), and its phosphorylation was detected with a BAS imager (top). Similar to HA-tagged HB-EGF, nontagged HB-EGF is phosphorylated by TPA stimulation. Immunoprecipitation of HB-EGF was confirmed by immunoblotting with anti-HB-EGF antibody (bottom). Arrowhead indicates a nonspecific band. (C) Phosphorylation of HB-EGF by various shedding stimuli. Vero cells expressing HA-tagged wild-type HB-EGF were incubated with [32P]-orthophosphate and stimulated with TPA (64 nM), LPA (20 μg/ml), anisomycin (Aniso, 1 μM), or ionomycin (Iono, 2 μM) in the presence of GM6001 for 30 min. Lanes indicated were pre-treated with PKC inhibitor Ro-31-8220 (10 μM). Cell lysates were immunoprecipitated with control IgG (–) or anti-HA antibody (+). Immunoprecipitated samples were subjected to SDS-PAGE and analyzed by a BAS imager (top right) or immunoblotting with anti-HA polyclonal antibody (bottom). Phosphorylated HB-EGF upon TPA stimulation is also shown at a shorter exposure time (top left). (D) Effect of PKC inhibitor on ectodomain shedding of HB-EGF. Vero cells expressing HA-tagged wild-type HB-EGF were stimulated with various shedding inducers for 30 min in the presence or absence of PKC inhibitor Ro-31-8220 (10 μM), and ectodomain shedding was detected by monitoring generation of the C-terminal fragment by immunoblotting using anti-HB-EGF C-terminus antibody (HB C-ter). Actin was used as a loading control. |
Ectodomain shedding of HB-EGF can be induced by various stimuli, including LPA, anisomycin, and ionomycin (Umata et al., 2001; Takenobu et al., 2003). Since TPA induces both ectodomain shedding and serine phosphorylation of HB-EGF, we next asked whether treatment of cells with other ectodomain shedding inducers causes HB-EGF phosphorylation. Vero cells expressing HA-tagged HB-EGF was metabolically labeled with [32P]-orthophosphate and stimulated with LPA, anisomycin, or ionomycin in the presence of GM6001, and then phosphorylation of proHB-EGF was examined. LPA, anisomycin, and ionomycin were able to induce phosphorylation of proHB-EGF, though at lower levels than TPA (Fig. 3C).
To test whether protein kinase C (PKC) might be involved in HB-EGF phosphorylation induced by these stimuli, we examined the effect of the PKC inhibitor Ro-31-8220. Ro-31-8220 significantly reduced phosphorylation of proHB-EGF caused by TPA and ionomycin treatment, but did not inhibit LPA- or anisomycin-induced phosphorylation. Under the same conditions, Ro-31-8220 inhibited TPA-induced ectodomain shedding (Fig. 3D), but not LPA-, anisomycin-, and ionomycin-induced ectodomain shedding. These results suggest that ectodomain shedding and the phosphorylation of HB-EGF are coordinated but separable processes, and that PKC activation is not required for HB-EGF phosphorylation.
We next examined whether HB-EGF phosphorylation is a general event by using another cell line, Buffalo rat liver (BRL) cells. We generated BRL cells expressing nontagged wild-type or S207A mutant HB-EGF. BRL cells metabolically labeled with [32P]-orthophosphate were stimulated with TPA in the presence of GM6001, and phosphorylation of HB-EGF was detected by immunoprecipitation with anti-HB-EGF antibody followed by autoradiography. Phosphorylation was strongly induced upon TPA stimulation (Fig. 4A), but weak phosphorylation of proHB-EGF was detected in serum-starved BRL cells. Phosphorylation of proHB-EGF was not detected in the S207A mutant with or without TPA stimulation.
 View Details | Fig. 4. HB-EGF phosphorylation in BRL cells. (A) HB-EGF is cell autonomously phosphorylated in BRL cells. BRL cells expressing nontagged wild-type or S207A mutant HB-EGF were incubated with [32P]-orthophosphate and stimulated with TPA in the presence of GM6001. Phosphorylation of HB-EGF was detected with a BAS imager. Heavily phosphorylated HB-EGF bands were detected from TPA-stimulated BRL cells, while weakly phosphorylated HB-EGF bands were detected from serum-starved nonstimulated BRL cells. Arrowhead indicates a nonspecific band. (B) Comparison of phosphorylation level between nontagged and HA-tagged HB-EGF under steady-state culture conditions. BRL cells expressing nontagged or HA-tagged HB-EGF were metabolically labeled in serum-starved conditions. Nontagged HB-EGF is phosphorylated in BRL cells, and the HA tag increases the phosphorylation level of HB-EGF. No phosphorylation was detected from the S207A mutant regardless of the presence or absence of an HA tag. Arrowhead indicates a nonspecific band. (C) Comparison of phosphorylation level between nontagged and HA-tagged HB-EGF under TPA stimulation. Nontagged HB-EGF is phosphorylated in BRL cells, and the phosphorylation level of HB-EGF is increased by the HA tag. No phosphorylation was detected from S207A mutant, even when cells were stimulated with TPA. |
We next addressed effect of the HA tag on Ser207 phosphorylation, because the phosphorylation level of HB-EGF appeared to be increased by the addition of the HA tag (Fig. 1A). We found that the phosphorylation level of HA-tagged HB-EGF was about 4.5 times higher than that of nontagged HB-EGF, when normalized to the levels observed in serum-starved cells (Fig. 4B). Moreover, the phosphorylation level of HA-tagged HB-EGF was about 1.5 times higher than that of nontagged HB-EGF when cells were stimulated with TPA (Fig. 4C). We could not detect any difference in phosphorylation level of nonstimulated cells in serum-starved or serum-fed culture conditions (data not shown). These results suggest that HB-EGF is phosphorylated in steady-state culture conditions, with no apparent cell-type specific effects. In addition, phosphorylation and dephosphorylation appears to be affected by adjacent amino acids, as suggested by differences observed between tagged and nontagged HB-EGF constructs.
In order to investigate the biological significance of C-terminal phosphorylation of HB-EGF, we examined the effect of phosphorylation on DT sensitivity. ProHB-EGF has been shown to act as a receptor for DT, which binds to proHB-EGF and is then internalized into endosomes. Thus, the sensitivity of cells to DT is affected both by the number of proHB-EGF molecules and the rate of internalization of proHB-EGF from the cell surface into endosomes. BRL cells expressing a nontagged or HA-tagged version of wild-type or S207A mutant HB-EGF were exposed to various concentrations of DT, and DT toxicity was determined by measuring inhibition of protein synthesis. Although the rate of protein synthesis inhibition was variable when cells were exposed to low concentrations of DT, no significant difference in DT sensitivity between cells expressing wild-type and S207A mutant HB-EGF was observed (Fig. 5A). This suggests that the cell surface concentration and internalization rate of proHB-EGF were not largely affected by the S207A mutation.
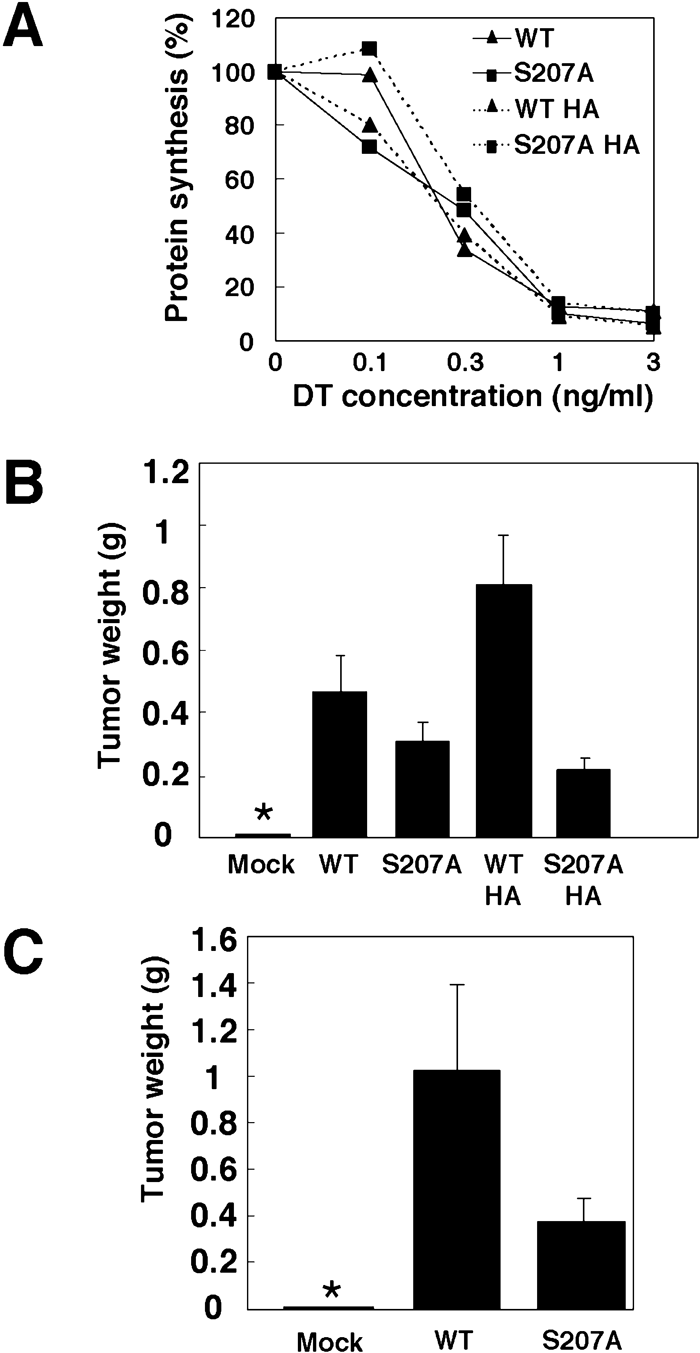 View Details | Fig. 5. S207A mutant has a reduced tumorigenic activity in nude mice. (A) DT sensitivity of BRL cells expressing wild-type or S207A mutant HB-EGF. BRL cells expressing nontagged or HA-tagged HB-EGF and S207A mutant HB-EGF were incubated with DT at the indicated concentrations, and the rate of protein synthesis was measured by incorporation of [3H]-leucine. (B) Tumorigenesis of BRL cells expressing various forms of HB-EGF. BRL cells expressing nontagged and HA-tagged wild-type or S207A mutant HB-EGF were injected into nude mice. Four weeks after injection, the weights of tumors were measured (n=5). Each bar indicates the mean (±SD). An asterisk indicates no measurable tumors. (C) Tumorigenesis of MEFs derived from an HB-EGF null mouse. The immortalized MEFs expressing nontagged wild-type or S207A mutant HB-EGF were injected into nude mice. Six weeks after injection, the weights of tumors were measured (n=5). Each bar indicates the mean (±SD). An asterisk indicates no measurable tumors. |
We previously reported that overexpression of HB-EGF promotes tumorigenesis of ovarian carcinoma cells (Miyamoto et al., 2004). To see whether the serine phosphorylation site is involved in HB-EGF-dependent growth of tumors, BRL cells expressing wild-type or S207A mutant HB-EGF were injected subcutaneously into nude mice. To avoid artifacts resulting from clonal variation of cells, nonclonal cells expressing similar amounts of wild-type, S207A mutant, or HA-tagged forms of proHB-EGF were used (Fig. 6). The S207A mutation reduced the tumorigenic activity of HB-EGF to 68.0% of wild-type (Fig. 5B, C). HA-tagged HB-EGF showed stronger tumorigenic activity than nontagged HB-EGF, and the Ser207 mutation abolished the enhanced tumorigenic activity. The differences in tumorigenic activities of these various forms of HB-EGF were correlated with phosphorylation level of each form of HB-EGF (Fig. 4B).
 View Details | Fig. 6. Surface expression and ectodomain shedding of wild-type and S207A mutant HB-EGF under steady-state culture conditions. (A) Western blot analysis. Total proHB-EGF (top), cell surface proHB-EGF (middle) and secreted soluble HB-EGF (bottom) were detected from whole cell lysates, HB-EGF monoclonal antibody precipitates of biotin-labeled cell lysates, and conditioned medium of BRL cells cultured for 48 h with serum-free medium, respectively. Actin is detected from total cell lysates. (B) DT binding assay for determining the cell surface level of HB-EGF. BRL cells expressing wild-type or S207A mutant HB-EGF were incubated with 125I-labeled DT and the specific binding was determined. Each bar indicates the triplicate mean (±SD). An asterisk indicates no DT binding. (C) Levels of secreted soluble HB-EGF as determined by DT binding assay. Soluble HB-EGF was enriched with heparin-Sepharose from the conditioned medium of BRL cells, and the amount of HB-EGF was measured by binding of 125I-labeled DT to heparin-Sepharose, as described in Materials and Methods. Each bar indicates the triplicate mean (±SD). (D) ERK activation by conditioned medium of BRL cells. Conditioned medium of control BRL cells (Mock), and BRL cells expressing HA-tagged wild-type or S207A mutant HB-EGF were prepared by culturing cells with serum-free medium for 48 hours. Serum-starved BRL cells were stimulated with serially diluted (10-fold dilutions) conditioned medium of control BRL cells (Mock) or BRL cells expressing HA-tagged wild-type (WTHA) or S207A mutant (S207AHA) HB-EGF for 10 min. PhosphoERK (top) and total ERK (bottom) were detected by immunoblotting. |
To further confirm the effect of phosphorylation on tumor forming activity and to exclude the effect of wild-type HB-EGF, which is endogenously expressed in the parental cells, we next performed similar experiments using immortalized MEF cells derived from an HB-EGF null mouse. The MEF cells were infected with retrovirus encoding wild-type or S207A mutant HB-EGF or with control mock retrovirus and stable lines were established. No difference was observed between the amount of wild-type and S207A mutant proHB-EGF expressed on the cell surface by surface biotinylation assay (data not shown). When these cells or mock-infected cells were injected subcutaneously into nude mice, cells expressing wild-type and S207A mutant HB-EGF, but not mock-infected cells, formed tumors (Fig. 5C). However, the tumors formed by MEF cells expressing wild-type HB-EGF were much larger than those formed by those expressing S207A mutant HB-EGF, suggesting that the tumorigenic activity of HB-EGF is markedly reduced by the phosphorylation deficient mutation.
A previous study indicated that ectodomain shedding is essential for HB-EGF-induced tumor formation (Miyamoto et al., 2004), while the present study showed that the S207A mutant of HB-EGF has reduced tumorigenic activity in BRL cells. These results led us to test the possibility that the S207A mutation affects ectodomain shedding of HB-EGF in BRL cells, even though TPA-induced shedding of HB-EGF was apparently not affected by the S207A mutation in Vero cells (Fig. 2B). We therefore examined ectodomain shedding of wild-type and S207A mutant proHB-EGF in BRL cells in steady-state conditions (constitutive cleavage). BRL cells expressing wild-type or S207A mutant HB-EGF were cultured in serum-free medium, and the levels of HB-EGF in total cell lysates, at the cell surface, and secreted into the conditioned medium were determined by immunoblotting. The amount of total and cell surface expression of proHB-EGF, as well as soluble HB-EGF in the condition media, were not influenced by the S207A mutation or by the HA tag (Fig. 6A). To quantitate HB-EGF levels in the conditioned media and on the cell surface, a 125I-labeled DT binding assay was used. Consistent with the results from the immunoblotting, cell surface expression and secretion of soluble HB-EGF were not affected by the S207A mutation or by the HA tag (Fig. 6B, C).
It is possible that the S207A mutation indirectly affects the mitogenic activity of soluble HB-EGF or that the C-terminal phosphorylation of HB-EGF induces secretion of other growth factors. Therefore, we examined the mitogenic activity of the conditioned medium of BRL cells expressing wild-type or S207A mutant HB-EGF. Conditioned medium of mock-infected BRL cells did not cause significant ERK phosphorylation of serum-starved BRL cells, while that of BRL cells expressing HA-tagged wild-type or S207A mutant HB-EGF caused ERK phosphorylation at similar levels (Fig. 6D). These results suggest that the S207A mutation does not influence the constitutive ectodomain shedding of HB-EGF, mitogenic activity of soluble HB-EGF, or production of other growth factors that activate ERK.
It has been reported that BAG-1 and PLZF bind to the cytoplasmic domain of HB-EGF to promote cell survival and growth (Nanba et al., 2003; Lin et al., 2001). Thus, it is possible that phosphorylation of HB-EGF is critical for their binding and activities. However, we could not detect BAG-1 or PLZF from HB-EGF immunoprecipitates of either nonstimulated or TPA-stimulated BRL cells (data not shown).
Finally, we examined whether other EGFR ligands could be phosphorylated by TPA stimulation. We established BRL cells stably expressing HA-tagged AR, β-cellulin (BTC), epiregulin (ER), or TGF-α. Cells were stimulated for 30 min in the presence of GM6001 and [32P]-orthophosphate, and HA-tagged EGFR ligands were immunoprecipitated with anti-HA antibody (Fig. 7). Phosphorylation of AR or ER was not detected. In contrast, like HB-EGF, TGF-α was phosphorylated by TPA stimulation. Interestingly, BTC was readily phosphorylated, and TPA stimulation did not change the level of phosphorylation. These results point to novel roles for phosphorylation in the modulation of biological activities of some EGF family growth factors.
 View Details | Fig. 7. TPA-induced phosphorylation of EGFR ligands. BRL cells expressing C-terminally HA-tagged HB-EGF (HB), amphiregulin (AR), β-cellulin (BTC), epiregulin (ER), or transforming growth factor-α (TGF-α) were stimulated with TPA in the presence of [32P]-orthophosphate and GM6001 for 30 min. Immunoprecipitates were subjected to SDS-PAGE and phosphorylation was detected with a BAS imager (top). Immunoblotting with anti-HA antibody shows bands for the different HA-tagged EGFR ligands (bottom). Phosphorylated forms are shown by asterisks. |
In this study, we were able to uncover novel features of the cytoplasmic domain of the EGF family of growth factors. First, we showed that the cytoplasmic domain of HB-EGF is phosphorylated by stimuli that are known to induce ectodomain shedding, and that phosphorylation occurs cell autonomously. Second, mutation of the phosphorylation site reduces HB-EGF-dependent tumor growth, and this effect is not due to reduced ectodomain shedding or cell surface proHB-EGF expression. Finally, other EGF family growth factors, including TGF-α and BTC, have potential phosphorylation sites. This is the first demonstration that the cytoplasmic domain of EGF family growth factors undergoes phosphorylation.
The activity of the EGF family of growth factors depends on the binding of receptor to the EGF-like domain of the ligand and the subsequent activation of the growth signaling cascade. Ectodomain shedding has been shown to be critical for regulating activity by controlling the release of the EGF-like domain into the extracellular space. We have previously shown that abrogation of ectodomain shedding of HB-EGF leads to abnormal development in mice and loss of tumorigenic activity (Miyamoto et al., 2004; Yamazaki et al., 2003). In addition, factors affecting the transport of the membrane-anchored precursor to the plasma membrane also contribute to regulated production of the soluble EGF-like domain. Our data show that a phosphorylation-deficient mutant of HB-EGF reduces tumor growth without affecting ectodomain shedding and trafficking of HB-EGF. Moreover, no difference was observed in the ability of wild-type and S207A mutant HB-EGF to induce ERK activation. These findings suggest that phosphorylation of HB-EGF is involved in the regulation of cell growth, but not via modulation of the canonical signaling pathway.
Ectodomain shedding is necessary for the tumorigenic activity of HB-EGF (Miyamoto et al., 2004). It is clear that the C-terminal phosphorylation site is not essential for ectodomain shedding, nor is ectodomain shedding required for phosphorylation. However, the two processes appear nonetheless to occur coordinately. For example, LPA is a potent inducer of ectodomain shedding of HB-EGF (Prenzel et al., 1999; Umata et al., 2001), and also induces the C-terminal phosphorylation of HB-EGF, as shown in this study. Thus, the phosphorylated C-terminal fragment of proHB-EGF might contribute to cell growth by cooperating with soluble HB-EGF upon ectodomain shedding.
It has been reported that the C-terminal fragment of HB-EGF that is generated by ectodomain shedding translocates to the nucleus and enhances cell cycle progression by inducing transcription of cyclin A through sequestration of the transcriptional repressor PLZF (Nanba et al., 2003). However, it has been reported that the ten C-terminal amino acid residues, including Ser207, are not required for the transcriptional activator function of the C-terminal fragment of HB-EGF. More recently, it has been shown that charged amino acid residues in the intracellular juxtamembrane region are important for transcriptional activation (Nanba et al., 2004). A previous study showed that BAG-1 binds to the cytoplasmic domain of HB-EGF, synergistically protecting cells from etoposide-induced apoptosis. In addition, binding of BAG-1 to HB-EGF increases secretion of HB-EGF (Lin et al., 2001). In our system, however, we could not detect binding of BAG-1 and PLZF to HB-EGF or any changes in the secretion level of the phosphorylation-deficient HB-EGF mutant. Therefore, phosphorylation site of HB-EGF may contribute to tumorigenesis through a novel mechanism that does not involve BAG-1 or PLZF.
It has been reported that tyrosine and serine/threonine kinases can be co-immunoprecipitated with proTGF-α (Shum et al., 1994). Interestingly, we found that TGF-α is also phosphorylated by TPA stimulation. It has also been shown that proHB-EGF and proTGF-α form complexes with membrane proteins such as CD9, integrin α3β1, and heparan sulfate proteoglycan (Nakamura et al., 1995; Shi et al., 2000). Thus, it is likely that kinases are recruited to the complexes and phosphorylate the cytoplasmic domain of HB-EGF upon external stimulation. Although the mechanism of cell growth affected by HB-EGF phosphorylation remains unknown, it is possible that the phosphorylated cytoplasmic domain of HB-EGF serves as a scaffold for signal transduction molecules and is involved in EGFR-independent signal transduction.
In conclusion, Ser207 of proHB-EGF is phosphorylated upon extracellular stimulation, and the S207A mutant has reduced tumor-forming activity. Further analysis of the molecular role and mechanism of HB-EGF phosphorylation on cell growth will be important for understanding this novel role of the HB-EGF cytoplasmic domain.
We thank Drs. T. Akagi, T. Kitamura, K. Miyazaki, M. Klagsbrun, and T. Sasazuki for providing cell lines, retrovirus vectors, and cDNA. This work was supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology (16207014 and 17014057 to E. M.) and by the Charitable Trust Osaka Cancer Research Fund (to H. M.).
|