| To whom correspondence should be addressed: Masamitsu Yamaguchi, Department of Applied Biology, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan. Tel: +81–75–724–7781, Fax: +81–75–724–7760 E-mail: myamaguc@kit.ac.jp Abbreviations: dScgβ, Drosophila β-sarcoglycan; DGC, dystrophin glycoprotein complex; LGMD, limb girdle muscular dystrophy; PI, propidium iodide. |
β-sarcoglycan is a member of the sarcoglycan complex. This forms part of the dystrophin glycoprotein complex (DGC) components, which have been seen to be mutated in muscular dystrophy (Ozawa et al., 2005). Sarcoglycan complexes are heteromeric, consisting of α-, β-, γ-, δ-, ε-, and ζ-sarcoglycans in humans (Ettinger et al., 1997; Lim et al., 1995; Nigro et al., 1996; Noguchi et al., 1995; Roberds et al., 1994; Wheeler et al., 2002). Abnormalities in the α-, β-, γ-, and δ-sarcoglycans genes are responsible for autosomal recessive limb girdle muscular dystrophies (LGMD2D-2F) (Ozawa et al., 2005; Vainzof et al., 1996), and mutations in the ε-sarcoglycan encoding gene are associated with myoclonus-dystonia syndrome of neurological origin (Zimprich et al., 2001).
One of the proposed functions of the sarcoglycan complex is to stabilize DGC itself by interacting with dystrophin, dystroglycan, and other constitutive proteins (Chen et al., 2006; Crosbie et al., 1999; Durbeej et al., 2000; Straub et al., 1998). The β-/δ-sarcoglycan core is known to interact with the C-terminus of dystrophin (Chen et al., 2006). Biochemical preparations from sarcoglycan mutant muscle have revealed less tightly adherent α-dystroglycan subunits suggesting abnormal interaction in the absence of sarcoglycan (Durbeej et al., 2000; Straub et al., 1998). Furthermore, lack of sarcoglycans has been demonstrated to cause instability of the DGC in several sarcoglycan-null mouse models (Durbeej and Campbell, 2002). Moreover, recent studies have indicated that sarcoglycans might also have non-mechanical functions, for example, functionally compensating for integrins in muscle (Allikian et al., 2004). α-Sarcoglycan has an ATP-binding site in the extracellular region, and α-sarcoglycan-transfected HEK293 cells exhibit a significant increase in ATP-hydrolyzing activity that is abolished by anti-α-sarcoglycan antibodies, suggesting the existence of ecto-ATPase activity (Betto et al., 1999; Sandona et al., 2004). It has also been reported that sarcoglycans associate with integrins and nNOS, indicating a potential role in signaling transduction. In fact, we have shown that β-sarcoglycan negatively regulates the Egfr signaling pathway (Hashimoto and Yamaguchi, 2006). These reports suggest that sarcoglycans are multifunctional proteins.
To date, the function of DGC in muscle has been extensively studied. However, despite current evidence that components of DGC are ubiquitously expressed in various mammalian tissues such as retina, brain, blood-brain barrier, and kidney (Chan et al., 2005; Culligan et al., 1998; Dalloz et al., 2001; Fort et al., 2005; Lien et al., 2006; Sekiguchi, 2005), roles in such non-muscle tissues remain obscure. Their elucidation should help in understanding DGC and the pathogenesis of muscular dystrophies.
To assess DGC functions, various animal models have been utilized. In addition to rodents such as mice, rats and hamster, C. elegans and zebrafish are currently used for research into human diseases (Bassett and Currie, 2003; Chamberlain and Benian, 2000). For instance, in the δ-sarcoglycan anti-sense morpholin-injected zebrafish, other associated sarcoglycans are downregulated, while dystrophin expression is not affected. A similar phenotype is typical of human patients carrying mutations in δ-sarcoglycan, suggesting that the zebrafish is an excellent model for the analysis of the limb-girdle muscular dystrophy (Guyon et al., 2005). Orthologs of some of the DGC members are also found in C. elegans (Grisoni et al., 2002)
Recently, Drosophila has also been utilized as an animal model to study human diseases (Celotto and Palladino, 2005; O’Kane, 2003). The majority of the mammalian DGC protein subclasses is well conserved in Drosophila (Greener and Roberts, 2000), but little is known whether their properties and functions are also evolutionary conserved. To address this question, we here investigated the expression pattern of Drosophila β-sarcoglycan (dScgβ) protein in various tissues and its subcellular localization. In the present report we document further evidence for the validity of Drosophila as a model animal in which to study limb-girdle muscular dystrophy, showing that the expression pattern of Drosophila β-sarcoglycan in various tissues is similar to that of mammalian β-sarcoglycan. Furthermore, dScgβ was found to localize in cytoplasm in addition to plasma membranes, with dramatic changes depending on the cell cycle.
Fly stocks were maintained at 25°C on standard food containing 0.7% agar, 5% glucose and 7% dry yeast. Canton S was used as the wild type. Transgenic flies carrying Act5C-GAL4>UAS-dscgβ and Act5C-GAL4>UAS-IR-dscgβ were established as described previously (Hashimoto and Yamaguchi, 2006).
Rabbit polyclonal anti-dScgβ antibodies were produced as described previously (Hashimoto and Yamaguchi, 2006), and used at 1:4,000 dilution for Western immunoblot analysis, and at 1:200 dilution for immunohistochemistry. Mouse monoclonal anti-α-tubulin and anti-lamin antibodies (Developmental Studies Hybridoma Bank) were both used at 1:400 dilution for immunohistochemistry, and anti-PCNA antibodies (Sigma) were used at 1:2,000 dilution for Western immunoblotting.
Third instar larval extracts from Canton S and transgenic flies carrying Act5C-GAL4>UAS-dscgβ and Act5C-GAL4>UAS-IR-dscgβ or embryonic extracts from Canton S were applied to SDS-polyacrylamide gels containing 7.5% or 10% acrylamide and proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad). The blotted membranes were blocked with TBS-T containing 2% bovine serum albumin, followed by incubation with the primary antibodies for 16 h at 4°C. After washing, the membranes were incubated with HRP conjugated secondary antibodies (Amersham Biosciences) at 1:5,000 dilution for 1 h at 25°C. Detection was performed with ECL Western blotting detection reagent and images were analyzed with a Lumivision Pro HSII image analyzer (Aisin Seiki).
Canton S embryos were collected, dechorionated in a 50% Clorox solution, washed with 0.7 M NaCl/0.2% Triton X-100 and fixed in a 1:1 heptane: 4% paraformaldehyde in TBS mixture for 20 min with vigorous shaking at 25°C. Vitelline membranes were then removed by shaking embryos in heptane-methanol at 25°C for 30 sec. Embryos were blocked with TBS containing 10% goat serum and 0.15% Triton X-100 (TBT) before incubation with the primary antibodies in the same solution for 16 h at 4°C. The embryos were washed extensively in TBS containing 0.3% Triton X-100, reblocked with TBT and incubated with the secondary antibody for 3 h at 25°C. After extensive washing with TBS containing 0.3% Triton X-100, they were mounted and observed using a Zeiss LSM510 confocal laser scanning microscope. For propidium iodide (PI) staining, embryos after fixation were treated with 200 μg/ml RNAase for 3 h at 37°C, washed extensively, and stained with 20 μg/ml PI for 2 h at 25°C. For phalloidin staining, embryos after fixation were incubated with 1 unit/200 μl phalloidin for 20 min at 25°C.
Third instar larvae of Canton S were dissected and the tissues were fixed with 4% paraformaldehyde in TBS mixture for 20 min at 25°C. The steps after fixation were the same as for the protocol for immunohistochemistry of embryos.
With antibodies against the full length dScgβ, two major bands could be detected on Western blots of Flag-dScgβ-over-expressed larval extracts (Fig. 1A, lane 1 and 2), with estimated molecular weights of 48 kDa and 114 kDa. These two bands could also be observed with anti-Flag antibodies, indicating that they are exogenously over-expressed dScgβ (Fig. 1A, lane 1 and 2). Based on amino acid sequence, dScgβ would be expected to migrate at 38,114. The slower migration observed here on SDS-PAGE is likely to be due to its unusual amino acid composition. The 114 kDa band may represent a dimeric or trimeric form of dScgβ. Dimer or multimer formation in the presence of SDS has been reported for mammalian sarcoglycans (Shi et al., 2004). It has also been reported that some cell surface antigens such as HLA-DR, -DC1 and -B7 form dimeric or multimeric forms even in the SDS-PAGE (Signas et al., 1982; Bono and Strominger, 1982). Anti-dScgβ antibodies could also detect a weak band at around 54 kDa in extracts from both Flag-dScgβ over-expressing and wild type larvae (Fig. 1A, lane 2 and 3). Moreover, this band was largely reduced in the larval extract from the dScgβ knock down flies (Fig. 1B). The data thus suggest that although this weak 54 kDa band is larger than expected, it does represent endogenous dScgβ. The discrepancy is likely to be due to some post-translational modification of dScgβ, such as phosphorylation and glycosylation, as reported for mammalian β-sarcoglycan (Shi et al., 2004).
 View Details | Fig. 1. Western immunoblot analysis. (A, B) Specificity of the affinity purified anti-dScgβ antibody. Proteins in extracts of third instar larvae of Act5C-GAL4>UAS-flag-dScgβ (lane 1 and 2) and Canton S (lane 3) were separated by SDS-polyacrylamide gel electrophoresis. Lane 1 was blotted with anti-Flag antibody and lanes 2 and 3 with anti-dScgβ antibody. White arrowheads indicate the exogenously overexpressed dScgβ and the black arrowhead the endogenous dScgβ. (B) Proteins in the extracts of third instar larvae of Act5C-GAL4>UAS-IR-dScgβ (lane 1) and Canton S (lane 2) were separated by SDS-polyacrylamide gel electrophoresis and blotted with anti-dScgβ antibodies. The black arrowhead indicates the endogenous dScgβ. Reduction of dScgβ protein in RNAi flies (lane 1) is evident, indicating that the 54 kDa band represents endogenous dScgβ. (C) Developmental Western blot during embryogenesis. Proteins in the extracts of unfertilized (lane 1), 0–4 h (lane 2), 4–8 h (lane 3), 8–12 h (lane 4), 12–16 h (lane 5), and 16–20 h (lane 6) embryos were separated by SDS-polyacrylamide gel electrophoresis and blotted with anti-dScgβ antibodies. Unfertilized embryos (lane 1) show maternal storage. In all experiments for this figure, PCNA was used as a total protein loading control. |
Developmental Western blot analysis during embryogenesis showed ubiquitous expression of dScgβ (Fig. 1C). Levels of dScgβ protein were highest in unfertilized embryo extracts, suggesting abundant maternal storage of dScgβ (Fig. 1C, lane 1). These data are consistent with strong anti-dScgβ immunostaining in ooplasm (Fig. 3C). Immunohistochemistry of the whole mount embryos showed dScgβ protein to be highly expressed in midgut (Fig. 2A) and the musculature, including dorsal and ventral oblique muscles (Fig. 2B), pleural muscles (Fig. 2B), and the dorsal pharyngeal musculature (Fig. 2A). In mammals, β-sarcoglycan is primarily expressed in skeletal muscle, smooth and cardiac muscle, in line with the fact that deficiency of β-sarcoglycan constitutes the primary cause of LGMD 2E (Bonnemann et al., 1996). The high expression of dScgβ protein in musculature is consistent with that of mammalian β-sarcoglycan.
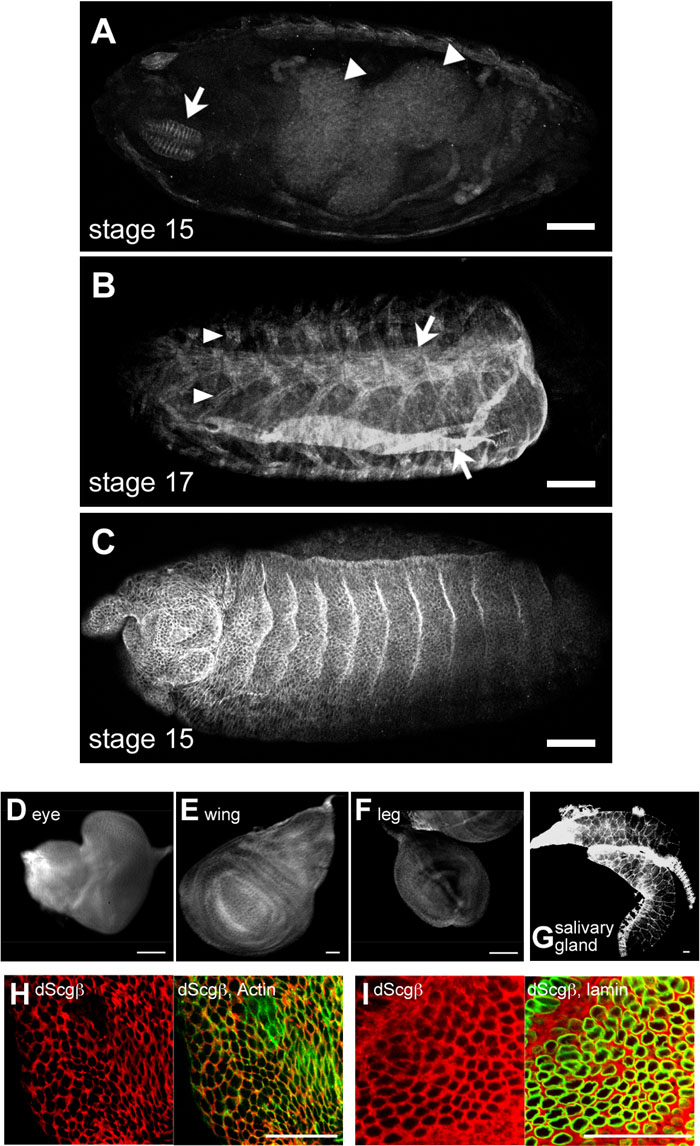 View Details | Fig. 2. Expression pattern of dScgβ during development. (A–C) whole mounts of staged embryos were stained with anti-dScgβ antibody. Arrowheads and arrows in panel A indicate the midgut and dorsal pharyngeal musculature, respectively. Arrowheads and arrows in panel B show pleural muscles and oblique muscles, respectively. dScgβ is also expressed in epithelial cells of embryos as shown in panel C. (A and B) dorsal view. (C) lateral view. (D–G) Imaginal discs and salivary gland are stained with anti-dScgβ antibodies. In all tissues tested here, anti-dScgβ signals were detected. (H, I) Enlarged images of eye imaginal discs. Anti-dScgβ signals are shown in left panels of H and I. The merged images of anti-dScgβ and phalloidin or anti-lamin signals are shown in right panels of H and I, respectively. dScgβ is localized in cytoplasm (panel I) in addition at plasma membrane (panel H). Bars in panel A–G indicate 100 μm, and bars in panels H and I indicate 20 μm. |
To date, embryonic expression patterns of Drosophila DGC component orthologs have been determined using RNA in situ hybridization (Dekkers et al., 2004). In the RNA level, dscgβ is expressed in brain, ventral nerve cord, and midgut (Dekkers et al., 2004). However, dScgβ protein was not detected in brain and ventral nerve cord (Fig. 2A). These differences can be explained by variation in the turnover rate of dScgβ protein in different tissues. In stage 17 embryos, the musculature is already differentiated, while the neural tissues such as brain and ventral nerve cord are still proliferating. In mammalian systems, it is reported that in proliferating rat C2C12 cultured myocytes, mRNAs for α-, β-, γ-, and δ-sarcoglycans are readily detectable while levels of proteins are very low, indicating a high turnover rate in the proliferating cells (Noguchi et al., 1999). Furthermore, it has also been demonstrated that accumulation of sarcoglycan proteins is accompanied by progress of C2C12 cell differentiation (Noguchi et al., 1999). Therefore, it is likely that the turnover rate of dScgβ is very rapid in proliferating cells like embryonic neural tissues but slow in differentiated cells such as the musculature in Drosophila.
Furthermore, we found that dScgβ is also expressed in epithelial cells of embryos (Fig. 2C), larval imaginal discs (Fig. 2D–F), and salivary glands (Fig. 2G). In mammals, sarcoglycan complexes are present not only in muscle but also in various non-muscle tissues such as the retina and endothelium (Claudepierre et al., 2000; Dalloz et al., 2001; Fort et al., 2005; Ramirez-Sanchez et al., 2005). Also in zebrafish, in situ hybridization analysis has shown that δ-sarcoglycan mRNA can be detected in brain and retina, in addition to the primary detection in the skeletal and cardiac muscles (Cheng et al., 2006). The varied expression of sarcoglycans is therefore conserved among species, suggesting multifunctional roles for sarcoglycans.
We therefore investigated the intercellular localization of dScgβ in eye imaginal discs by double staining with anti-dScgβ antibody and phalloidin to visualize plasma membrane by binding filamentous actin, or anti-lamin antibodies to visualize nuclear membranes. We found dScgβ to be localized both in plasma membranes (Fig. 2H) and in cytoplasm (Fig. 2I). Previous immunohistochemistry data for myotube cultures from normal human muscle showed an intercellular localization for β-sarcoglycan at early stages of myoblast fusion. It subsequently shifts to many spots in the perinuclear area, and finally localizes at the sarcolemma (Radojevic et al., 2000). Thus dScgβ may have a role not only on plasma membranes but also in the cytoplasm.
The predominant localization of mammalian DGC components in plasma membranes is well known. On the other hand, several recent studies have revealed that mammalian DGC components are also present in cytoplasm and nuclei (Calderilla-Barbosa et al., 2006; Fuentes-Mera et al., 2006; Marquez et al., 2003; Radojevic et al., 2000). However, no study which addresses their dynamics of distribution on cell cycle progression has been reported so far.
Drosophila provides examples of many distinct cell cycle patterns all in a single organism. During embryogenesis, the first 13 cycles show nearly synchronous nuclear division consisting of S and M phases. After cellularization, most epidermal cells undergo three more cell cycles (cycles 14–16) that are regulated by S/G2/M transition. Following mitosis 16, most embryonic cells enter an extended G1 phase. Some of the G1-arrested cells such as imaginal discs that give rise to the adult body resume G1/S/G2/M cell cycles only after the larva hatches. Other G1-arrested cells subsequently resume rounds of endoreplication (S/G cycle), giving rise to polyploid or polytene larval cells such as cells in the salivary glands.
Firstly we examined the dScgβ distribution in embryos before cellularization. dScgβ was detected in the whole embryo, suggesting maternal storage in the ooplasm (Fig. 3C). In cycle 14, cellularization starts with S/G2/M type regulation, as described above, and group of cells, termed mitotic domains, enter mitosis in close synchrony with each other, but out of synchrony with cells of other mitotic domains. The anti-dScgβ staining data revealed a non-plasma membrane localization of dScgβ in mitotic domains (Fig. 3D). PI staining allowed us to distinguish each phase of the cell cycle easily. Intriguingly, we found that the subcellular distribution of dScgβ is dramatically changed dependent on the cell cycle in embryos (Fig. 4A). In S phase cells, dScgβ is localized in the cytoplasm and plasma membranes (Fig. 4A). In M phase cells, it moves away from the membranes, appearing to associate with the ends of the chromosomes, as if regulating chromosomes segregation and cytokinesis (Fig. 4A). The anti-dScgβ staining pattern in M phase cells is very similar to that of tubulin. Therefore, we carried out double staining of embryos with anti-dScgβ and anti-α-tubulin antibodies. The data showed at least partial co-localization of the two (Fig. 4B). In embryos before cellularization, the high background makes it difficult to detect any localization change of dScgβ during M phase.
 View Details | Fig. 3. Expression pattern of dScgβ during early embryonic development. (A) Normalski image of (B). (B) Negative staining control without first antibody. No anti-dScgβ signals being observed. (C) Whole mount of early stage embryos stained with anti-dScgβ antibody. Early embryos before cellularization (S/M cycle) appear to have abundant maternal storage of dScgβ protein. (D) Whole mount of stage 6 embryo stained with anti-dScgβ antibody. Broken-line circles show examples of mitotic domains after cellularization (S/G2/M cycle) where anti-dScgβ signals at plasma membranes are diminished. All bars indicate 100 μm. |
 View Details | Fig. 4. Sub-cellular distribution of dScgβ during the cell cycle. (A and B) Enlarged images of cells in mitotic domains (S/G2/M cycle) are shown. Red and green indicate PI and anti-dScgβ signals, respectively in panel A, while in panel B, red and green indicate anti-tubulin and anti-dScgβ signals, respectively. Arrowheads in panel B show examples of partial co-localization between dScgβ and tubulin. (C) An eye imaginal disc (G1/S/G2/M cycle) double stained with anti-tubulin (red) and anti-dScgβ antibodies (green). The merged image is shown in the right panel. Arrowheads in panel E show examples of cells undergoing mitosis. Partial co-localization of dScgβ and tubulin was observed also in eye imaginal discs. (D) Salivary gland (S/G cycle) double stained with anti-dScgβ antibodies (red) and phalloidin (green). The merged image is shown in the right panel. Arrows indicate plasma membrane localization of dScgβ while arrowheads show perinuclear localization. A reticulated pattern in the cytoplasm is also evident. Bars in panel A, panel B, panel C, and panel D indicate 2,5 μm, 10 μm, 5 μm, and 20 μm, respectively. |
We next examined the behavior of dScgβ in eye imaginal discs of third instar larvae (Fig. 4C), where typical G1/S/G2/M type cell cycles prevail. As observed in embryos, dScgβ was localized in cytoplasm in addition to plasma membranes during S phase (Fig. 4C). Again, it was also seen to be partially co-localized with tubulin during M phase (Fig. 4C), suggesting that the dynamic change of dScgβ localization during mitosis appears to be common among different tissues. It is well known that DGC closely interacts with the actin cytoskeleton through dystrophin. However, an interaction with tubulin has not been previously reported. The present work is the first report suggesting their possible interaction.
Finally we examined the salivary glands where cells are regulated by S/G type cell cycling. dScgβ was predominantly localized at plasma membranes, as observed with other tissues (Fig. 4D), with a reticulated pattern in the cytoplasm and perinuclear localization also being clearly observed. In addition, dScgβ localization in the nuclear matrix was also detected (Fig. 4D).
In S phase cells of all tissues tested here, we observed cytoplasmic distribution of dScgβ in addition to localization at plasma membranes (Fig. 4). Furthermore, at least partial co-localization of dScgβ and tubulin was observed in M phase cells. These data taken together suggest that dScgβ might play a positive role in onset or progression of mitosis in association with tubulin. It is interesting to examine the effect of dScgβ knock down on the progression of the cell cycle. However, it has so far proved to be technically difficult to knock down dScgβ in proliferating cells and tissues, most likely because of the relatively long half-life of the dScgβ protein and a high abundance of dScgβ mRNA in these cells (Hashimoto and Yamaguchi, 2006). Establishment and analysis of dScgβ mutant fly lines may help to clarify this point. The presence of β-sarcoglycan in nuclei was previously reported in HeLa cells based on confocal microscopy and also its association with the nuclear matrix. Similar observations have also been made for Dp71 dystrophin, β-dystroglycan, α- and β-syntrophin, α1- and β-dystrobrevin (Fuentes-Mera et al., 2006). Moreover, Dp71 dystrophin is also localized in the nuclei of PC12 cells (Calderilla-Barbosa et al., 2006). However, the roles of these proteins in nuclei remain to be elucidated. In the present study, we found that dScgβ changes its localization coupled with cell cycle progression. We also suggest that the movement of dScgβ during mitosis may due to the association with tubulin. Our findings point to further functions of some DGC components in nuclei.
We are grateful to Drs M. Moore and S. Cotterill for comments on the English manuscript. This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
|