| To whom correspondence should be addressed: Kaori Tsutsumi, Department of Health Sciences, School of Medicine, Hokkaido University, N12-W5, Kita-ku, Sapporo 060-0812, Japan. TEL & FAX: +81–11–706–3421 E-mail address: tsutsumi@cme.hokudai.ac.jp |
Non-small cell lung cancer (NSCLC) is one of the most common causes of mortality in Japan and elsewhere, accounting for about 80% of all lung cancer cases. Approximately 37% of such patients will present with stage IIIA or IIIB advanced disease (Jemal et al., 2006). Despite recent developments in radiotherapeutic techniques, such as intensity-modulated radiation therapy (IMRT) and 3-dimensional conformal therapy, treatment of advanced NSCLC results in 5-year survival rates of only 10–20% (Spira and Ettinger, 2004). This is partly because most patients present with advanced disease, rendering tumoricidal doses of irradiation difficult to apply. Although radiotherapy together with platinum-based chemotherapy slightly improves the survival of patients with stage IV and inoperable stage III NSCLC compared with radiation alone (Spira and Ettinger, 2004), the survival advantage is only 10–15%.
The p53 gene is mutated in over 50% of NSCLCs (Ahrendt et al., 2000), which gene contributes to irradiation-induced apoptosis. In fact, p53 status is a prognostic factor for radiotherapy and chemotherapy (Bristow et al., 1996; Ohnishi et al., 2002).
Radiotherapy and chemotherapy for the treatment of malignant tumors exploit the cell responses such as G1 arrest, DNA repair and apoptosis that are induced by radiation and DNA damaging agents. However, the mechanisms of these responses remain unclear.
Cellular damage by irradiation or drugs results from breaks in single or double DNA strands. Double strand breaks are repaired by non-homologous end joining (NHEJ) or homologous recombination (HR), and these mechanisms as well as their importance to genomic instability have started to be investigated in detail. Proteins belonging to the RecA/Rad51 family promote the early steps of recombination in all organisms (West, 2003).
Recent studies have indicated that Rad51 paralogs play significant roles in recombination. The RAD51 paralogs, RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3, are involved in homologous recombination in mammalian cells and are required for normal levels of resistance to DNA-damaging agents, particularly cross-linking drugs, such as cisplatin and mitomycin (Thacker, 1999; Tarsounas et al., 2004). Tarsounas et al. (2004) reported that RAD51D, of which the sequence is approximately 39% similar to RAD51, is also involved in telomere maintenance (Tarsounas et al., 2004). Furthermore, rad51d-deficient mouse cells exhibit extensive chromosome instability, centrosome fragmentation, and do not form radiation-induced RAD51-foci (Smiraldo et al., 2005). RAD51D is important for both DNA repair and telomere maintenance, and is therefore essential for cell viability. In addition, deletion mutants of rad51 paralogs including rad51d in chicken DT40 cells that harbor a p53 defect are highly sensitive to cisplatin compared with wild type cells (Yonetani et al., 2005). Similarly, Smiraldo et al. (2005) reported that double mutants of rad51d and p53 are more sensitive to DNA-damaging agent than p53 single mutants.
The present study examines the radio- and chemoresistance of the p53-null NSCLC cell line, H1299. An interesting chemosensitivity was observed for H1299-IR, which survived 10 Gy irradiation (H1299-IR). Basal levels of mRNA expressions were compared between the parental H1299 and H1299-IR cells. In this report, we discuss the possible mechanisms of the chemosensitivity of H1299-IR.
The human non-small cell lung cancer cell lines, H1299 and A549, were obtained from ATCC. Both lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, GIBCO/Invitrogen Co., Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Sigma, St. Louis, MO, USA) and 50 μg/ml Kanamycin (Wako Pure Chemical Industries Ltd., Tokyo, Japan) at 37°C in a humidified 95% air –5% CO2 atmosphere.
Semi-confluent cultures, approximately 1×106 cells, were exposed to 10 Gy of X-rays at room temperature using a linear accelerator (Mitsubishi Electric Co., Tokyo, Japan). The cells were immediately dispersed with trypsin and roughly 3×105 cells were spread onto culture dishes (100 mm diameter). After 14 days, all colonies (approximately 1×103 colonies), were harvested and we referred to them as H1299-IR cells.
Total RNA extracted from semi-confluent parental H1299 cells and from H1299-IR cells was labeled and hybridized onto a human microarray chip. Detected signals were then examined using computer analysis (Agilent Technologies, Palo Alto, CA, USA).
Cells that survived X-ray irradiation were analyzed using a clonogenic assay. Viable tumor cells (1×104–105 cells) were seeded onto 60-mm dishes and cultured in DMEM containing 10% FBS. After 10 to 14 days, cells were fixed with methanol and stained with 2% Giemsa solution (Kanto Chemical Co. Inc., Tokyo, Japan) to determine the number of colonies per dish. Values were corrected by comparison with the plating efficiency of the untreated cells.
The numbers of apoptotic cells were determined by Annexin V staining followed by FACS analysis. Cells were incubated with 25 μg/ml cisplatin for 48 h, washed with ice cold PBS and adjusted to a density of 1×106 cells/ml. Annexin V binding was assayed using the Annexin V: FITC Apoptosis Detection Kit I (BD Biosciences Pharmingen, San Jose, CA, USA). We detected DNA fragmentation as follows. Genomic DNA isolated from semi-confluent cultures exposed to 25 μg/ml cisplatin for 48 h was separated by 2% agarose gel electrophoresis. All basic DNA manipulations proceeded as described by Sambrook et al. (1989).
Cells were seeded into 96-well plates at a density of 5×103 cells/well, incubated overnight and then exposed to 25 μg/ml cisplatin for 2 h. At the indicated time, MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] and phenazine ethosulfate was added to each well and after 40 min at 37°C, the absorbance of each well was measured at 492 nm. All experiments were performed in triplicate. Background absorbance was determined by incubating medium with substrate alone and subtracting the values from wells containing cells. The MTS assay was performed using the CellTiter Aqueous OneSolution kit (Promega Corporation, Madison, WI, USA).
To inhibit apoptosis, the pan-caspase inhibitor Z-VAD-FMK (100 μM; Peptide Institute, Inc., Osaka, Japan) was mixed with the reaction medium for cisplatin treatment.
After 10 Gy irradiation with 6 MV X-rays, we harvested all surviving cell colonies and we referred to this population as H1299-IR. To determine the difference in radiosensitivity between parental H1299 and H1299-IR, we assayed colony formation after irradiation with 10 Gy. The effects upon each cell line did not significantly differ. In contrast, 10 Gy was lethal to the p53 wild-type NSCLC cell line, A549 (Fig. 1). The parental H1299 and H1299-IR cell lines were about 100-fold more resistant to X-ray exposure.
 View Details | Fig. 1. Effect of radiation on parental H1299, H1299-IR and A549 cells. After 10 Gy radiation, cells were seeded in 60-mm dishes and colony formation was assayed. Cell viability was determined from the ratios (%) of viable cells compared with total numbers of seeded cells. Values represent means (±SD) of duplicate experiments. |
The growth of H1299-IR was similar to that of the parental cell line (Fig. 2).
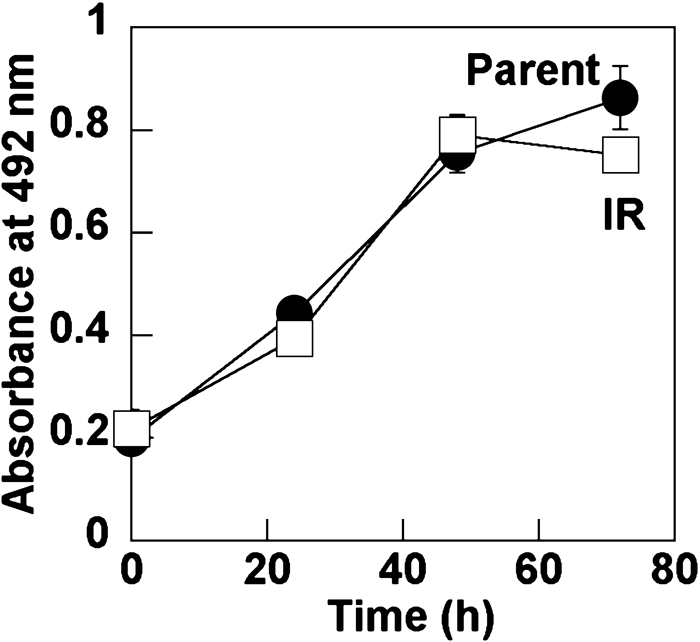 View Details | Fig. 2. Comparison of H1299 and H1299-IR cell growth. Wells of 96-well plates were seeded with 5,000 cells each and growth was monitored using MTS assays. Closed and open symbols indicate H1299 (Parent) and H1299-IR (IR) cells, respectively. Values represent means (±SD) of triplicate experiments. |
Since cisplatin is most commonly used reagent for NSCLCs, we investigated the chemosensitivity of the H1299-IR cell line to 25 μg/ml of this drug. The growth of parental cells was slightly repressed after 2 h of exposure to cisplatin compared with untreated cells, but maintained until 48 h. However, the MTS activities of H1299-IR cells were significantly reduced within 24 h. H1299-IR cells were about 2-fold more sensitive than parental cells both at 24 and 48 h according to the MTS assay (Fig. 3A). This trend was confirmed by an experiment similarly using another H1299-IR (data not shown). The cell morphology differed between H1299 and H1299-IR cells after incubation with cisplatin (Fig. 3B). The H1299-IR cells were round and smaller than the parental cells.
 View Details | Fig. 3. Effect of cisplatin on cell viability. (A) Wells of 96-well plates were seeded with 5,000 each of H1299 (closed symbols, Parent) and H1299-IR (open symbols, IR) cells and 24 h later, 25 μg/ml cisplatin was added for 2 h. Cell viability was monitored by MTS assays. Values represent means (±SD) of triplicate experiments. (B) Semiconfluent H1299 and H1299-IR cells were incubated with cisplatin (25 μg/ml) for 72 h. Morphology of H1299 (Parent) and H1299-IR (IR) cells was monitored by light microscopy. |
To identify the pathway of H1299-IR cell death after exposure to cisplatin, we performed the MTS assay in the presence of the pan-caspase inhibitor, Z-VAD-FMK. H1299-IR cell death induced by cisplatin was blocked by Z-VAD-FMK (Fig. 4A). These results suggested that caspase-dependent apoptosis was the predominant mode of death. In addition, FACS analysis showed that the number of apoptotic H1299-IR cells was about 2-fold higher than that of parental cells (Fig. 4B). We then separated the genomic DNA of both cell lines by electrophoresis on 2.0% agarose gels. Degradation of genomic DNA was detected as a smear pattern in H1299-IR cells, whereas neither a smear pattern nor ladder formation was evident for the parental cells (Fig. 4C).
 View Details | Fig. 4. Cisplatin-induced apoptosis in H1299 and H1299-IR cells. (A) Wells of 96-well plates were seeded with 5,000 each of H1299 (Parent) and H1299-IR (IR) cells and 24 h later, 25 μg/ml cisplatin was added for 48 h. Cell viability was monitored using MTS assays. To inhibit the caspase-dependent apoptosis pathway, cells were treated with 100 μM Z-VAD-FMK (open columns) or left untreated (closed columns). Values represent means (±SD) of triplicate experiments. (B) H1299 and H1299-IR cells (2.5×105 each) were seeded in 60-mm dishes and incubated for 24 h before adding cisplatin (25 μg/ml) for 48 h. Apoptotic cells were discriminated by annexin V staining followed by FACS analysis. (C) Cisplatin-induced DNA degradation. H1299 and H1299-IR cells (1×106 each) were seeded on 100-mm dishes and incubated for 24 h. Cisplatin (25 μg/ml) was applied for 48 h and genomic DNA was isolated. Fragmentation of DNA was detected by 2.0% agarose gel electrophoresis. |
Basal levels of gene expression were compared by micro-array analysis (Agilent) between parental H1299 and H1299-IR (Table I). Among 30,000 genes, 351 and 368 genes were up-and down-regulated in H1299-IR, respectively. Twelve and 15 genes were significantly up and down regulated, respectively (up to 2-fold). In particular, rad51d, a homologous recombination repair protein was down-regulated 2.8-fold and matrix metalloproteinase 1 (MMP1) was up-regulated 4.4-fold.
H1299-IR cells, which survived 10 Gy irradiation, became more sensitive to cisplatin but maintained tolerance to ionizing irradiation. H1299-IR cells exposed to cisplatin underwent apoptosis that was detected by both annexin V staining and DNA fragmentation on electrophoresis compared with the parental line. The morphological image of H1299-IR cells differed from that of H1299 after exposure to cisplatin. These cisplatin-induced apoptosis were blocked by Z-VAD-FMK. However, the growth curves of parental H1299 and H1299-IR did not significantly differ.
Smirado et al. (2005) have shown that p53- and rad51d-double defective mouse embryonic fibroblasts are cisplatin-sensitive. Yonetani et al. (2005) also recently reported that rad51d-deficient chicken DT40 cells are extremely sensitive to cisplatin and that both wild type and rad51d-deficient cells are similarly radioresistant. Their study used p53-defective DT40 cells. Our findings were consistent with their results, since H1299-IR cells, in which rad51d is down-regulated, were chemosensitive, but radiation tolerant.
Table I shows that 12 and 15 genes were significantly up- and down-regulated, respectively. Apart from rad51d, matrix metalloproteinase 1 (MMP1), also known as collaganase-1 gene, was the most up-regulated gene (4.4-fold increase). Since MMP1 is related to tumor invasion, growth, migration and metastasis (Stamenkovic, 2003; Brinckerhoff et al., 2000), H1299-IR might have aggressive properties, such as local invasion or distant metastasis. We found that MMP1 was also up-regulated in the A549-IR lung adenocarcinoma cell line, which is p53 wild type (data not shown). The trend was similar in a mouse fibrosarcoma cell line (data not shown). These findings indicate that X-irradiation universally up-regulates MMPs.
The clinical implication of our findings is that p53-deficient NSCLCs become sensitive to cisplatin after a dose of X-irradiation in the order of 10 Gy. Notably, solitary primary lung tumors measuring up to 3 cm can be successfully treated with 12 Gy doses of X-rays up to a total of 48 Gy (Onishi et al., 2004). However, a slight decrease of fraction size, such as to 10 Gy, up to a total of 40 Gy does not control the primary tumor (Onishi et al., 2004). Later regimens are primarily applied to tumors located at the hilum because of concerns about the large arteries. Our results suggest that these tumors are suitable for high-dose cisplatin therapy with concomitant irradiation since cells that survived 10 Gy irradiation became sensitive to chemotherapy.
Patients with mutations in p53 have a significantly higher risk for lung cancer-related death (Skaug et al., 2000) and preclinical data show that p53 status might be able to predict the sensitivity of lung cancer to radiation or chemotherapy (Viktorsson et al., 2005). In fact the present study found that p53-null H1299 cells were more tolerant to radiation than A549 cells.
Furthermore, the present findings support the notion that cisplatin (at concentrations in the order of 10–20 μg/ml), either together with, or after radiotherapy is an effective strategy for the treatment of p53-null lung cancers. Administering a high dose of cisplatin by conventional intravenous injection is not easy, but intra-arterial administration has proven feasible for treating head-and-neck cancers, in which p53 is mutated in about 50% of patients (Boyle et al., 1993; Brennan et al., 1995). The results of intra-arterial infusions of drugs combined with radiotherapy have also been quite impressive (Robbins et al., 2005; Homma et al., 2005).
Since MMP1 gene expression was elevated in H1299-IR cells (which probably have more invasive and metastatic potential), cells that survive X-ray radiation must be eradicated. High-dose cisplatin injection directly into tumors seems a very attractive option.
Our results are still preliminary, and further investigations are required to understand the mechanisms of the increased cisplatin sensitivity in the p53-null cell line. Such studies should reveal the molecular mechanisms of radiation- and chemosensitivity of lung cancers. Another implication from our study is that tumors with a similar gene profile to H1299-IR before treatment might be successfully treated with cisplatin either concurrently with or subsequent to X-ray radiotherapy.
This study was supported by a Grant-in-Aid for Scientific Research (B18390325) provided by the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Ms. Rie Yamazaki for excellent technical assistance with the X-ray irradiation and Dr. Yusuke Ohba for critical reading of this manuscript and valuable discussion.
|