| To whom correspondence should be addressed: Hiroshi Ohno, Laboratory for Epithelial Immunobiology, Research Center for Allergy and Immunology, RIKEN, 1-7-22 Suehiro, Tsurumi, Yokohama, Kanagawa 230-0045, Japan. Tel: +81–45–503–7031, Fax: +81–45–503–7030 E-mail: ohno@rcai.riken.jp Abbreviations: Alexa 633-Tfn, Alexa Fluor 633-conjugated human transferrin; di-RFP, tandem-dimer red fluorescent protein; EEA1, early endosome antigen1; EM, electron microscopy; FBS, fetal bovine serum; GFP, green fluorescent protein; KFP1, kindling fluorescent protein 1; mAb, mouse monoclonal antibody; PB, sodium phosphate buffer; PBS, phosphate-buffered saline; ROI, region of interest; TfR, transferrin receptor; TGN, trans-Golgi network; PA-GFP, photoactivatable GFP; VSV-G, vesicular stomatitis virus glycoprotein G. |
Eukaryotic cells have an elaborate internal membrane system that collectively comprises the biosynthetic/secretory and endocytic pathways. The trans-Golgi network (TGN) plays a pivotal role in sorting newly synthesized membrane proteins in the biosynthetic pathway to appropriate cellular destinations (Palade, 1975; Traub and Kornfeld, 1997). Plasma membrane proteins are segregated into a distinct transport carrier bound for the cell surface. This TGN-to-plasma membrane route is commonly referred to as the constitutive pathway. Whereas substantial progress has been made in the identification and characterization of the sorting signals and the molecular mechanisms of intracellular protein trafficking in recent years, the precise movements and dynamics of plasma membrane proteins in the TGN-to-plasma membrane trafficking remain to be clarified.
The green fluorescent protein (GFP) is extensively used as an in vivo reporter for gene expression and intracellular protein trafficking in intact cells and organisms (Tsien, 1998). The subsequent development of fluorescence photobleaching techniques, such as fluorescence recovery after photobleaching and fluorescence loss in photobleaching, has aided studies of the kinetic properties of GFP-tagged proteins. In those techniques, a selected pool of fluorescent proteins in a given subcellular region is photobleached temporarily or repeatedly, and the kinetic parameters of the proteins are characterized by the change in fluorescence intensity within or outside the photobleached region (Axelrod et al., 1976; Reits and Neefjes, 2001; Lippincott-Schwartz et al., 2003). The fluorescence photoactivation technique is another promising approach to the study of protein kinetics (Yokoe and Meyer, 1996; Elowitz et al., 1997; Sawin and Nurse, 1997; Marchant et al., 2001). After irradiation with light having a different wavelength from that for imaging, photoactivatable fluorescent proteins display increased fluorescence that directly highlights a distinct pool of proteins within a cell and enables direct monitoring of the behavior of the given protein pool.
Recently, three photoactivatable fluorescent proteins, photoactivatable GFP (PA-GFP) (Patterson and Lippincott-Schwartz, 2002), Kaede (Ando et al., 2002) and kindling fluorescent protein 1 (KFP1) (Chudakov et al., 2003), were reported to display ≥30-fold increase in fluorescence after photoactivation. PA-GFP was developed based on the conventional photoconversion mode of Aequorea victoria wild-type GFP (wtGFP) (Prasher et al., 1992). wtGFP normally exists as a mixed population of neutral phenols and anionic phenolates that produce major and minor absorbance peaks at 397 nm and 475 nm, respectively. The photoconversion of wtGFP, which involves shifting from the neutral phenols predominantly to the anionic phenolates, is thought to take place upon intense irradiation with ultraviolet or ~400-nm light, giving rise to an increase in absorbance of the minor peak at 475 nm. PA-GFP, a wtGFP mutant in which Thr-203 is substituted with His, was found to have negligible absorbance of the minor peak but an approximately 100-fold increase in fluorescence when excited with 488-nm light after intense irradiation with 413-nm light (Patterson and Lippincott-Schwartz, 2002). Compared with Kaede and KFP1, the inherent brightness of GFP and the low self-association property of PA-GFP enable its use as a reliable fluorescent fusion tag to study protein localization and trafficking (Chalfie et al., 1994; Tsien, 1998; Zacharias et al., 2002).
In this study, first, we characterized the advantages of photoactivating PA-GFP with multiphoton laser light over that with single-photon laser light. Compared with the single-photon laser light, the multiphoton laser light showed stably high photoactivation efficiency and high spatial resolution in photoactivating PA-GFP in vivo. The multiphoton photoactivation technique was then applied to study the dynamic trafficking route of the newly synthesized human transferrin receptor (TfR) in non-polarized mammalian cells. By photoactivating the TfR tagged with PA-GFP (PA-GFP-TfR) at the TGN using two-photon 790-nm laser light and subsequent excitation for time-lapse imaging in living cells, we provided evidence that Tfn+EEA1+ sorting endosomes (Widera et al., 2003; Maxfield and McGraw, 2004) could serve as an intermediate compartment for TfRs after exiting the TGN.
The pPA-GFP-N1 mammalian expression plasmid was constructed by introducing four mutations (L64F, T65S, V163A and T203H) into WEGFP of pEGFP-N1 (BD Biosciences Clontech, Mountain View, CA) using polymerase chain reaction (PCR)-based site-directed mutagenesis as previously described (Patterson and Lippincott-Schwartz, 2002).
cDNA encoding the human TfR amplified from pcDNA3-TfR (Sugimoto et al., 2002) using primers 5'-GGAATTCTATGATGGATCAAGCTAGATCAG-3' and 5'-GGGATCCTTAAAACTCATTGTCAATGTCCC-3' was subcloned into pEGFP-C1 (BD Biosciences Clontech) to generate pEGFP-TfR. PA-GFP was amplified using primer 5'-GTCAGATCCGCTAGCGCTACCGGTCGCCAC-3' containing the Nhe I site and primer 5'-GGAATTCGAAGCTTCAGCTCGAGATCTGAGTCCGGACTTGTACAGCTCGTCCATGCCGAGAGTGATCC-3' containing the EcoR I site, using pPA-GFP-N1 as the template. The fragment was digested with restriction endonucleases Nhe I and EcoR I, and ligated to the similarly digested pEGFP-TfR to produce pPA-GFP-TfR.
cDNA encoding a tandem-dimer red fluorescent protein (di-RFP) (a kind gift from Dr. Roger Y. Tsien, Howard Hughes Medical Institute, University of California at San Diego) was subcloned into pcDNA3 flanked by BamH I and EcoR I sites to generate di-RFP-pcDNA3. cDNA encoding the GRIP domain of rat Golgin-97 was amplified using primer 5'-GCGGAATTCGGGAGAAGACAGGCCCTG-3' containing the EcoR I site and primer 5'-CCGCTCGAGCTAGGACCATGGTATCCGAG-3' containing the Xho I site, and digested with restriction endonucleases EcoR I and Xho I. The fragment was ligated into similarly digested di-RFP-pcDNA3 to produce di-RFP-GRIP.
HeLa cells were grown in DMEM containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (10% DMEM) (all from Sigma-Aldrich). Cells grown on coverslips or glass-bottom dishes were transfected 36–48 hr before experiments using FuGENE 6 Transfection Reagent (Roche Molecular Biochemicals, Indianapolis, IL) following the manufacturer’s protocols.
Alexa Fluor 633-conjugated human transferrin (Alexa 633-Tfn) was from Molecular Probes (Eugene, OR). Cells were incubated with 20 μg/ml Alexa 633-Tfn in 10% DMEM for 10 min at 37°C. For immunofluorescence microscopy, coverslip-grown cells were fixed with 3% paraformaldehyde/phosphate-buffered saline (PBS), pH 7.4, for 15 min at room temperature after washing once with PBS. For live cell photoactivation experiments, cells grown on glass-bottom dishes were washed twice with 37°C pre-warmed 10% DMEM and used in photoactivation and time-lapse imaging as described in the section Photoactivation, imaging and analysis.
To confirm the localization of di-RFP-GRIP at the TGN, cells co-transfected with di-RFP-GRIP and PA-GFP-TfR were fixed and immunostained with a mouse monoclonal antibody (mAb) specific for human Golgin-97 (Molecular Probes, Eugene, OR) followed by an Alexa Fluor 488-conjugated goat anti-mouse IgG antibody (Molecular Probes, Eugene, OR). In some experiments, the cells grown on glass-bottom dishes were photoactivated as described in the section Photoactivation, imaging and analysis, incubated at 37°C for 1 min, then fixed and immunostained with a mouse mAb against early endosome antigen1 (EEA1) (BD Transduction Laboratories) followed by an Alexa Fluor 633-conjugated goat anti-mouse IgG antibody (Molecular Probes, Eugene, OR).
Single-photon photoactivation was performed with the 405-nm line of a diode laser (Coherent, Santa Clara, CA). Multiphoton photoactivation was performed with the 790-nm line of a Ti: sapphire laser (Mai Tai, Spectra-Physics, Mountain View, CA). For live cell experiments, the cells were grown on glass-bottom dishes, photoactivated and imaged on a Leica TCS SP2 AOBS confocal laser scanning microscope (Leica Microsystems Heidelberg GmbH, Wetalar, Germany) fitted with an optional live cell stage heater and a CO2 chamber at 37°C with 5% CO2. Up to 100 frames of xy-sections with 2-sec time interval after photoactivation were consecutively imaged to monitor the horizontal movements of activated fluorescent molecules. Photoactivation and time-lapse imaging were performed under the control of Advanced TimeLapse software. The regions of interest (ROIs), the power level of laser light, and the durations for photoactivation were defined at the bleach dialog window. In some experiments, the live cells were photoactivated with 790-nm two-photon laser light, incubated at 37°C with 5% CO2 for 1 min, and then fixed with 3% paraformaldehyde/PBS solution. After being immunostained with the antibodies indicated, the photoactivated cells were imaged in the xyz-mode under the indicated sequential excitations. The photoactivated region of the cells was further analyzed with Image J software. Quantification of fluorescence intensity and the number of pixels in the photoactivated regions of a fixed specimen was performed with Leica TCS SP2 system software.
Localization of diRFP-GRIP was analyzed immunoelectron microscopically using the pre-embedding gold enhancement method. HeLa cells cultured on plastic coverslips (LF, Sumitomo Bakelite, Tokyo, Japan) were transfected with diRFP-GRIP, and fixed with 4% paraformaldehyde (Nacalai Tesque, Kyoto, Japan) in 0.1 M sodium phosphate buffer (PB), pH 7.4, for 2 hr. After washing with the same buffer three times for 5 min, the fixed cells were incubated in PB containing 14% glycerol and 35% sucrose for 15 sec, and permeabilized by freezing and thawing in liquid nitrogen. The cells were washed with PB, blocked by incubating for 30 min in PB containing 0.005% saponin, 10% BSA, 10% normal goat serum, and 0.1% cold water fish skin gelatin, and exposed overnight to anti-RFP rabbit polyclonal antibody (MBL, Nagoya, Japan) (1/500 dilution) in the blocking solution. After washing with PB containing 0.005% saponin, the cells were incubated with colloidal gold (1.4-nm diameter, Nanoprobes Inc., NY)-conjugated goat anti-rabbit IgG in the blocking solution for 2 hr. The cells were then washed with PB and fixed with 1% glutaraldehyde in PB for 10 min. After washing with PBS containing 50 mM glycine, with PBS containing 1% BSA, and with milliQ water, gold labeling was intensified with a gold enhancement kit (GoldEnhance EM, Nanoprobes Inc., NY) for 5 min at room temperature according to the manufacturer’s instructions. After washing with distilled water, the cells were post-fixed in 1% OsO4 containing 1.5% potassium ferrocyanide in PB for 60 min at room temperature, and washed with distilled water. The cells were dehydrated in a series of graded ethanol solutions and embedded in epoxy resin. After the epoxy resin hardened, the plastic cover slip was removed from it. Ultra-thin sections were cut horizontally to the cell layer and doubly stained with uranyl acetate and lead citrate. Samples were analyzed with an H7600 electron microscope (Hitachi, Tokyo, Japan).
Recent developments in PA-GFP have enabled studies of intracellular protein dynamics by photolabeling photoactivatable molecules in a selected population and then monitoring the induced GFP fluorescence within the cell (Patterson and Lippincott-Schwartz, 2002). To determine the optimal conditions for photoactivation in vivo, we first examined the effect of power level and duration of 405-nm laser light on photoactivating PA-GFP expressed in HeLa cells (Fig. 1). Various conditions for photoactivation were applied to the same cell in which PA-GFP was expressed and appeared to distribute uniformly throughout the cytoplasm (Fig. S1, and data not shown). ROIs for photoactivation were defined at the bleach dialog window under the control of Leica Advanced TimeLapse software, allowing the smallest number of pixels to be selected for fluorescence induction. Both the fluorescence intensity and the number of pixels photo activated were power-dependent (Fig. 1A and B).
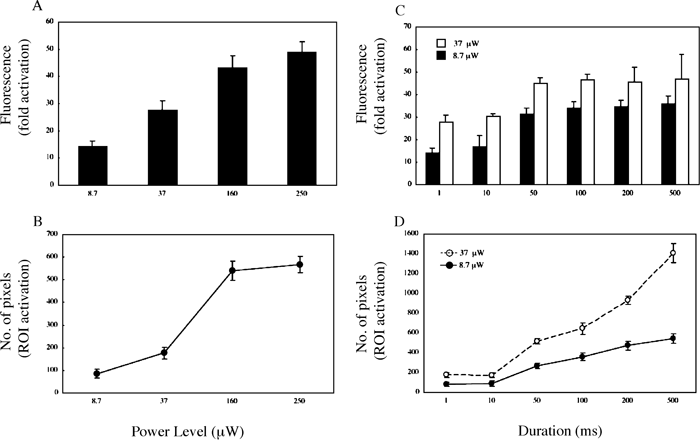 View Details | Fig. 1. Photoactivation with single-photon (405 nm) laser light. See Figure S1 for the representative image after photoactivation. PA-GFP was expressed in HeLa cells and imaged on xy-sections under 488-nm light excitation after irradiation with 405-nm laser light. (A and C) Quantification of fluorescence enhancement and (B and D) number of pixels in the photoactivated regions induced by (A and B) indicated power level (μW) for 1 ms or (C and D) 8.7 or 37 μW for indicated durations. The indicated power levels of the laser light were measured at the back aperture of the objective. Each point is derived from at least three separate experiments; error bars represent SD. |
 View Details | Fig. S1. Photoactivation of PA-GFP in vivo with single-photon (405 nm) laser light. PA-GFP was expressed in HeLa cells and imaged on xy-sections under 488-nm light excitation after irradiation with 405-nm laser light of 8.7, 37, 160, 250 μW for 1 ms (ROI 1~4), 8.7 μW for 10, 50, 100, 200, 500 ms (ROI 5~9), or 37 μW for 10, 50, 100, 200, 500 ms (ROI 10~14), respectively. The cell outline is marked by a dotted line. Definition of ROIs for photoactivation was performed with Advanced TimeLapse software through a Leica HCX PL APO CS 63×1.4 objective. A representative image is shown. Bar, 8 μm. |
To avoid the harmful effects of intense irradiation with near-ultraviolet light of ~400 nm on live cells, it is necessary to use the lowest power of UV light and the shortest duration. Under our experimental system, a 30~40-fold increase in fluorescence was obtainable after irradiation with 405-nm laser light of 8.7 μW for ≥50 ms (Fig. 1C) in the region of ≥200 pixels (Fig. 1D). Although ≥40-fold increase in fluorescence was induced with the power of 37 μW (Fig. 1C), much broader regions (≥500 pixels) were photoactivated (Fig. 1D). These data suggest that although a high power level or a long exposure time for ~400-nm laser excitation may achieve the maximum increase in fluorescence after photoactivation, this results in excessive photoactivation outside the ROIs and is unfavorable to live cells.
In order to confine the fluorescence induction to the vicinity of the probed region and prevent UV photodamage of live cells, we used multiphoton laser light to photoactivate PA-GFP in vivo. Multiphoton excitation involves the simultaneous absorption of two or more pulses of separate photons that collectively excite probe molecules to the excited state. The electronic transition occurs in a very small region near the focal point of the laser light (Denk et al., 1990; Denk 1994) and the red light of the multiphoton laser light is less harmful to live cells than the single-photon ~400-nm laser light used to photoactivate PA-GFP (Mohanty et al., 2002; Post et al., 2005). Under our experimental system, successful photoactivation of PA-GFP expressed in HeLa cells was achieved between 780 to 840 nm, with the highest fluorescence intensity observed at 790 nm under the maximum output (~600 mW) (data not shown).
Photoactivation efficiency and the extent of activation in three-dimensional (3D) space were further compared by using 405-nm laser light of 8.7 μW and 790-nm laser light of ~600 mW to photoactivate PA-GFP expressed in the same HeLa cells (Fig. S2 and Fig. 2). In contrast to using the 405-nm laser light, use of the 790-nm laser light showed duration-independent activated fluorescence intensities and defined regions on the focal plane for photoactivation (Fig. 2A and B). Exposure to 790-nm laser light for as short as 1 ms sufficiently produced 30~40-fold fluorescence enhancement that was restricted to 70~90 pixels on the focal plane, whereas ≥50 ms duration was required to obtain ≥30-fold fluorescence enhancement with photoactivation of ≥200 pixels in the case of 405 nm irradiation (Fig. 2A and B). Furthermore, confinement of the fluorescence of photoactivated PA-GFP excited along the laser light axis by 790-nm laser light was 2~2.5-fold higher than that by 405-nm laser light (Fig. 2C).
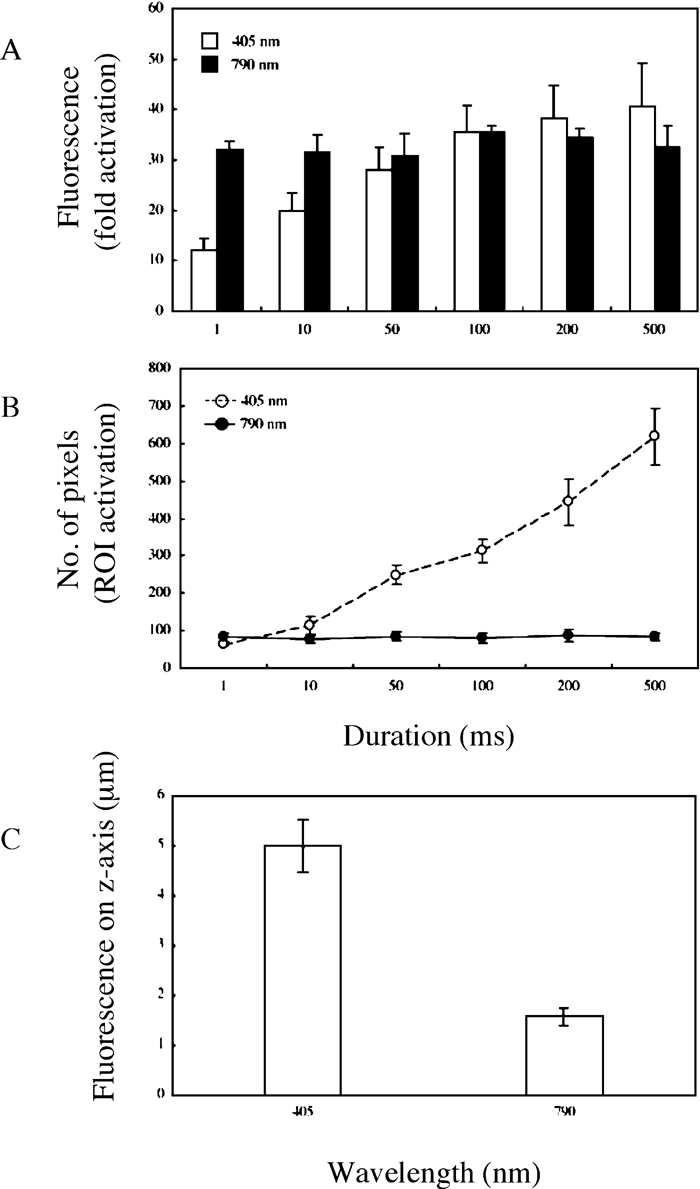 View Details | Fig. 2. Comparison of photoactivation with single-photon (405 nm) and multiphoton (790 nm) laser light. See Figure S2 for the representative image after photoactivation. PA-GFP was expressed in HeLa cells and imaged on xyz-sections under 488 nm light excitation after irradiation with either 405- or 790-nm laser light. (A) Quantification of fluorescence enhancement and (B) number of pixels in the photoactivated regions on the focal frame induced by irradiation with either 405-nm light of 8.7 μW or 790-nm light of 600 mW for indicated durations. (C) Quantification of photoactivated fluorescence on the z-axis induced by irradiation with 405-nm light of 8.7 μW or 790-nm light of 600 mW for 100 ms. Each point is derived from at least three separate experiments; error bars represent SD. |
 View Details | Fig. S2. Comparison of photoactivation of PA-GFP in vivo with single-photon (405 nm) and multiphoton (790 nm) laser light. (A) PA-GFP was expressed in HeLa cells and imaged on xy-sections under 488-nm light excitation after irradiation with 405-nm laser light of 8.7 μW (ROI 1~6) or 790-nm laser light of 600 mW (ROI 7~12) for 1, 10, 50, 100, 200, 500 ms, respectively. The cell outline is marked by a dotted line. (B and C) The same cell was imaged on xz-sections under 488-nm light excitation after irradiation with 405-nm laser light (B) of 8.7 μW or 790-nm laser light (C) of 600 mW for 50 ms. Definition of ROIs for photoactivation was performed with Advanced TimeLapse software through a Leica HCX PL APO CS 63×1.4 objective for 405-nm light and a Leica HCX PL APO CS 63×1.32 objective for 790-nm light. Representative images are shown. Bar, 8 μm. |
Taken together, these data suggest that the multiphoton laser light exhibited significantly higher efficiency and accuracy in photoactivating PA-GFP than the single-photon laser light.
Next, we applied the multiphoton photoactivation technique to study the intracellular transport of membrane proteins. It is known that newly synthesized TfRs are delivered from the Golgi apparatus to the plasma membrane where they are constitutively endocytosed and recycled via early endosomes back to the plasma membrane. However, the precise trafficking route of newly synthesized TfRs from TGN to the plasma membrane has not been fully elucidated; thus, it is of interest to ask whether early endosomal compartments are involved in the biosynthetic pathway. To address this issue, we needed to selectively photoactivate a pool of PA-GFP-TfRs at the TGN site and directly monitor the movement of the photoactivated PA-GFP-TfRs in living cells by time-lapse microscopy. To this end, the localization of the TGN must be visualized in living cells.
Golgin-97 is a peripheral membrane protein localized at the TGN and the GRIP domain at its C-terminus is essential for its localization (Munro and Nichols, 1999; Yoshino et al., 2003). We assumed, therefore, that the chimeric protein di-RFP-GRIP, in which the GRIP domain of Golgin-97 is fused with a tandem-dimer red fluorescent protein (di-RFP) (Campbell et al., 2002), should associate with the TGN membrane to serve as a live TGN marker. Indeed, we observed a good co-localization of di-RFP-GRIP with Golgin-97 (Fig. 3A–C), but not with transferrin-labeled endosomal compartments (Fig. 3D–F). Immunoelectron microscopy also showed that di-RFP-GRIP predominantly localized at the TGN, further confirming the TGN localization of di-RFP-GRIP (Fig. 3G).
 View Details | Fig. 3. Expression and localization of di-RFP-GRIP in HeLa cells. (A to F) HeLa cells were transfected to co-express di-RFP-GRIP and PA-GFP-TfR and examined by fluorescence microscopy or (G) EM. (A to C) The cells were fixed and immunostained with mAb against Golgin-97 followed by Alexa 488-conjugated goat anti-mouse antibody. (D to F) The cells were allowed to take up Alexa 633-Tfn into early endosomes before fixation and imaged under (A and D) 543-nm light excitation (red), (B) 488-nm light excitation (green) or (E) 633-nm light excitation (blue). C and F are merged images of A and B, and D and E, respectively. Bar, 8 μm. (G) Cells with moderate expression of di-RFP-GRIP were fixed and examined by EM as described in Materials and Methods. Large and small arrows indicate trans-sites of the Golgi apparatus and clathrin-coated vesicles, respectively. Arrowheads indicate endoplasmic reticulum (ER). Bar, 500 nm. |
Since PA-GFP-TfR exists abundantly in the endosomes (Fig. S3), similar to endogenous TfR, this raises the question whether the photoactivation of PA-GFP-TfR at the TGN concomitantly activates PA-GFP-TfR in the endosomal compartment. To examine whether TGN-restricted photoactivation of PA-GFP-TfR could be achieved, first, HeLa cells co-expressing di-RFP-GRIP and PA-GFP-TfR were allowed to internalize Alexa 633-Tfn before fixation. Then, multiphoton photoactivation was performed using 790-nm laser light at the TGN and the early endosomes, which were marked by di-RFP-GRIP and Alexa 633-Tfn, respectively.
 View Details | Fig. S3. Photoactivation of PA-GFP-TfR chimeric protein with single-photon (405 nm) laser light. The cells were prepared and imaged before (A to B) and after (D to F) photoactivation as described in Figure 4. Photoactivation was performed with 405-nm laser light of 8.7 μW for 10 ms and 100 ms in ROI1 and ROI2, respectively. Imaging and definition of ROIs for photoactivation (C) were performed with Advanced TimeLapse software using a Leica HCX PL APO CS 63×1.4 objective. White dots indicated by arrowheads denote the ROIs at the TGN sites defined for photoactivation. Bar, 8 μm. Red, green and blue fluorescence indicates di-RFP-GRIP, photoactivated PA-GFP-TfR and Alexa 633-Tfn, respectively. |
PA-GFP-TfR photoactivated at the TGN was restricted to the di-RFP-GRIP-labeled structure and did not overlap with the Alexa 633-Tfn-positive endosomal compartment (Fig. 4 and Video 1). In contrast, PA-GFP-TfR localized in areas beyond the TGN region for photoactivation, including Tfn-containing endosomal compartment and even possibly the plasma membrane, was photoactivated by the 405-nm laser light (Fig. S3). This would pose a problem in investigating the dynamic behavior of PA-GFP-TfR from one compartment to another. Taken together, the PA-GFP chimeric proteins at a restricted intracellular compartment could be selectively photolabeled by multiphoton laser light, enabling the direct monitoring of kinetic movements of the highlighted molecules in live cells.
 View Details | Fig. 4. TGN-restricted photoactivation of PA-GFP-TfR with multiphoton (790 nm) laser light. HeLa cells co-transfected with PA-GFP and di-RFP-GRIP were allowed to take up Alexa 633-Tfn into early endosomes and then fixed. After photoactivation with 790-nm laser light 600 mW for 1 ms at the TGN sites marked by di-RFP-GRIP, the cell was imaged in the xyz-mode under sequential excitation with 488-, 543- and 633-nm laser lights. Stacks in the squared region were further analyzed with the Image J software and shown as supplemental Video 1. Red, green and blue fluorescence indicates di-RFP-GRIP, photoactivated PA-GFP-TfR and Alexa 633-Tfn, respectively. White dots indicated by arrowheads denote the ROIs at the TGN sites defined for photoactivation. Bar, 8 μm. |
The multiphoton photoactivation technique was used to explore the intracellular trafficking pathway of PA-GFP-TfR from the TGN to the plasma membrane in live cells. HeLa cells co-expressing PA-GFP-TfR and di-RFP-GRIP were grown on glass-bottom dishes and loaded with Alexa 633-Tfn to mark the endosomes. Little fluorescence was seen before photoactivation (Fig. 5A and 5B), and no co-localization of di-RFP-GRIP and Alexa 633-Tfn was observed (Fig. 5C). PA-GFP-TfRs at the TGN site were probed with di-RFP-GRIP and defined for photoactivation (Fig. 5D). After irradiation with 790-nm laser light for 100 ms, bright fluorescence signals were seen to co-localize with the di-RFP-GRIP-marked TGN site (Fig. 5E) but not Alexa 633-Tfn loaded endosomes (Fig. 5F and 5G top left panel). Time-lapse imaging revealed that the photoactivated fluorescence signals of PA-GFP-TfR moved into the Tfn-labeled early endosomal compartment within 60–90 sec (Video 2 and Fig. 5G).
 View Details | Fig. 5. TfRs are transported from TGN to early endosomes in HeLa cells. A HeLa cell co-expressing PA-GFP-TfR and di-RFP-GRIP was loaded with Alexa 633-Tfn to mark early endosomes. The cell was imaged on xy-sections under indicated excitation laser light(s) before (A to C) and after (E to F) photoactivation. Arrows and arrowheads in E and F indicate the photoactivated fluorescent molecules. (D) Imaging and definition of ROIs for photoactivation with 790-nm laser light of 600 mW for 100 ms at TGN sites marked by di-RFP-GRIP were performed with the Advanced TimeLapse software through a Leica HCX PL APO CS 63×1.32 objective. Red, green and blue fluorescence indicates di-RFP-GRIP, photoactivated PA-GFP-TfR and Alexa 633-Tfn, respectively. White dots indicated by arrowheads denote the ROIs at the TGN sites defined for photoactivation. Bar, 8 μm. (G) The squared region (shown in F) from representative images of supplemental Video 2 are magnified and shown. Arrows and arrowheads denote the induced fluorescent molecules and the targeted early endosome, respectively. |
To confirm the movement of PA-GFP-TfR into the early endosomal compartment after exiting TGN, similar multiphoton photoactivation at the TGN was performed (Fig. 6A) followed by further incubation for 1 min to allow the induced fluorescence signals to exit the initial photoactivation site. The cells were then fixed and analyzed under a confocal immunofluorescence microscope. The fluorescence signals co-localized with the Tfn-containing endosomal compartment (Fig. 6B and 6C), suggesting that the newly synthesized TfR is transported from the TGN to the plasma membrane via endosomes.
 View Details | Fig. 6. TfRs are transported to Tfn-containing compartments in HeLa cells. (A) A HeLa cell co-expressing PA-GFP-TfR and di-RFP-GRIP was loaded with Alexa 633-Tfn and then photoactivated with 790-nm laser light of 600 mW for 100 ms at TGN sites marked by di-RFP-GRIP. Red, green and blue fluorescence indicates di-RFP-GRIP, photoactivated PA-GFP-TfR and Alexa 633-Tfn, respectively. White dots indicated by arrowheads denote the ROIs at the TGN sites defined for photoactivation. (B) One minute after photoactivation, the cell was fixed and the photoactivated cell was imaged in the xyz-mode under sequential excitation with 488- and 633-nm laser lights. The merged image of sections after fixation is shown. Bar, 8 μm. (C) Magnification of squared region in B. |
To further characterize the nature of the Tfn-containing early endosomal compartment containing the activated PA-GFP-TfR from the TGN, a cell co-expressing di-RFP-GRIP and PA-GFP-TfR was immunostained with a mAb against EEA1, a marker for sorting endosomes (Widera et al., 2003; Maxfield and McGraw, 2004), 1 min after multiphoton activation. The photoactivated GFP fluorescence of PA-GFP-TfR was detected in the EEA1-positive compartment (Fig. 7), which is most likely the sorting endosome (Sheff et al., 2002; Widera et al., 2003; Maxfield and McGraw, 2004).
 View Details | Fig. 7. TfRs are transported to EEA1-positive sorting endosomes from the TGN in HeLa cells. A HeLa cell co-expressing PA-GFP-TfR and di-RFP-GRIP was photoactivated with 790-nm laser light of 600 mW for 100 ms at TGN sites marked by di-RFP-GRIP. One minute after photoactivation, the cell was fixed and immunostained with mAb against EEA1 followed by Alexa 633-Tfn-conjugated goat anti-mouse antibody. The photoactivated cell was imaged in the xyz-mode under sequential excitation with 488- and 633-nm laser lights. Images of the xy-section containing induced fluorescent molecules under excitation with 488-nm (A) or, 633-nm (B) laser light, and the merged image (C) after fixation are shown. Red, green and blue fluorescence indicates di-RFP-GRIP, photoactivated PA-GFP-TfR and EEA1, respectively. Squared region in A to C are magnified and shown on the right top. Bar, 8 μm. |
Taken together, the time-lapse imaging of living cells and immunofluorescence microscopic analysis demonstrated that PA-GFP-TfR could be precisely photoactivated by multiphoton laser light and that upon TGN exit, the photoactivated PA-GFP-TfR are transported possibly directly to the Tfn+EEA1+ sorting endosomes.
Newly synthesized membrane proteins are sorted at the TGN for delivery to various cellular destinations including the plasma membrane, the endosomes and the lysosomes. Whereas much progress has been made in recent years to understand the sorting signals for and the molecular machineries of transmembrane protein trafficking in the biosynthetic/secretory pathway, the precise dynamic movements during the transport en route to their final destinations are less clear. This is largely because the transmembrane proteins are being continually synthesized and distributed; the investigation of such a dynamic system would be difficult, if not impossible, by biochemical methods and conventional microscopy. In this report, first, we established an efficient multiphoton photoactivation technique in which the PA-GFP fused chimeric proteins could be selectively photoactivated within a confined intracellular compartment. The precise movements of the activated fluorescent chimeric proteins were then tracked directly by time-lapse microscopy.
Recent developments in photoactivation using photoactivatable fluorescent proteins, such as the PA-GFP, Kaede and KFP1, combined with kinetic microscopy techniques provide a promising and straightforward approach to study the dynamics of protein transport in living cells. All of these molecules display low fluorescence at the excitation wavelength for imaging but significantly increase their fluorescence after a brief pulse of high-intensity irradiation at a different wavelength. For example, PA-GFP exhibited up to 100-fold increase in fluorescence when excited at 488 nm after irradiation with ~400-nm light (Patterson and Lippincott-Schwartz, 2002). When the same conditions were used for the photoactivation of Kaede (Ando et al., 2002), a remarkable ~2000-fold increase in its red-to-green fluorescence ratio was achieved. KFP1 uses green light at 532 nm for photoactivation, giving rise to ~30-fold increase in red fluorescence (Chudakov et al., 2003). However, the self-association of Kaede and KFP1 to form tetramers limits their usefulness as protein reporters. In contrast, the low self-association of PA-GFP (Zacharias et al., 2002) enables it to be used as a reliable fluorescent protein tag in live cells.
The optimal conditions for photoactivating PA-GFP in vivo were firstly examined in HeLa cells expressing PA-GFP by using 405-nm laser light of various power levels and durations (Fig. 1 and Fig. S1). Although the fluorescence of PA-GFP was increased with increasing power level and longer exposure time, the photodamage due to high-intensity near-ultraviolet light and the photoactivation extended areas are unfavorable for studies of temporal and spatial dynamic protein trafficking in live cells. This prompted us to seek an alternative excitation source for photoactivation instead of the ~400-nm laser light.
Two- or multiphoton excitation has been successfully used to image various fluorescent proteins, including GFP and its variants (Niswender et al., 1995; Zipfel et al., 2003). Because of its nature, multiphoton laser activates fluorescent molecules only in the focal plane (Denk et al., 1990), in contrast to the photoactivation of fluorescent proteins along the trajectory of the light in the case of the single-photon laser. In addition, the multiphoton laser uses a longer wavelength range than the single-photon laser for excitation of fluorescent molecules, which is less harmful to the cells. These characteristics of the multiphoton laser might be suitable for the photoactivation of PA-GFP. In general, fluorescent molecules can be excited by near-infrared multiphoton light whose wavelength is roughly twice that required for one-photon excitation. Therefore, it is reasonable to speculate that PA-GFP could be photoactivated using the ~800-nm wavelength of the multiphoton laser. As expected, GFP fluorescence could be successfully induced by the multiphoton laser light at the highest power level available in our microscope system for each wavelength ranging from 780 nm to 840 nm, with the highest fluorescence intensity induced by 790-nm laser light (data not shown). Since the output power at 790 nm was the highest among the other wavelengths tested (data not shown), the fluorescence enhancement is likely to be dependent on the power level of the multiphoton laser light. The fluorescence intensity of PA-GFP was increased by ~30- to 40-fold on irradiation with 790-nm laser light for as short as 1 ms, and the multiphoton photoactivation efficiency appeared to be independent of the duration (Fig. 2A). More importantly, the extent of photoactivation by 790-nm laser light was more highly restricted to the focal point on the focal plane of the laser light in 3D space compared with that by the single-photon laser light (Fig. S2, Fig. 2B and C), consistent with the phenomenon that the simultaneous absorption of separate photons for excitation occurs within the diffraction-limited focal spot (Denk et al., 1990). Recently, the two-photon photoactivation of a PA-GFP-fusion protein in live Drosophila embryos was found to maintain viability of the organism, whereas the cells irradiated by the single-photon laser light failed to divide properly in subsequent cell cycles (Post et al., 2005), suggesting the probability of phototoxicity of the single-photon activation of PA-GFP in mammalian cells. Taken together, these data support the notion that multiphoton photoactivation is more favorable for use to selectively photolabel PA-GFP within a sub-cellular location in living cells than single-photon activation.
In order to apply the multiphoton photoactivation technique to study the transport of proteins out of a given compartment in living cells, the compartment must be labeled in intact cells. We were interested in studying the dynamic movement of the newly synthesized PA-GFP-TfR from the TGN. To locate the TGN in living cells, we took advantage of the chimeric protein di-RFP-GRIP. This protein localizes at the TGN because of the GRIP domain of Golgin-97 that can mediate specific interaction with the TGN membrane (Munro and Nichols, 1999; Yoshino et al., 2003) (Fig. 3). By using di-RFP-GRIP to mark the TGN, the precise photoactivation of PA-GFP-TfR at the TGN has been successfully achieved by multiphoton photoactivation in both fixed and live HeLa cells (Fig. 4 and Fig. 5, respectively, compared with Fig. S3 and Video 3 by single-photon activation).
Earlier studies involving cell fractionation and electron microscopy (EM) analyses have provided evidence that the newly synthesized TfR and vesicular stomatitis virus glycoprotein G (VSV-G) could be detected in early endosomes prior to appearing on the surface of non-polarized mammalian cells (Hedman et al., 1987; Futter et al., 1995). More recently, the newly synthesized VSV-G was found to enter transferrin-positive endosomes after exiting from TGN in TfR-transfected Madin-Darby canine kidney cells (Ang et al., 2004). These studies suggest a pathway for some, if not all, of the newly synthesized plasma membrane proteins from the TGN to the early endosomes en route to the cell surface. Consistent with this notion, we observed that, after being photoactivated at the TGN site, the induced fluorescence of PA-GFP-TfR merged with fluorescent Tfn labeled-early endosomes in cultured HeLa cells (Fig. 5, Fig. 6 and Video 2).
Early endosomes can be further subdivided into two distinct organelles: EEA1+rab11– sorting endosomes and EEA1–rab11+ recycling endosomes (Widera et al., 2003; Maxfield and McGraw, 2004). The destination of PA-GFP-TfR after exiting the TGN was also positive for EEA1 (Fig. 7), suggesting that the photoactivated PA-GFP-TfR is probably delivered directly to the sorting endosomes after exiting from the TGN. We cannot formally exclude the possibility that PA-GFP-TfR is first delivered to the other compartments, including the recycling endosomes, before appearing in the sorting endosome. Nevertheless, we prefer the former possibility, since the timing of the arrival of the photoactivated PA-GFP to the Tfn-labeled compartment and the EEA1-labeled compartment is similar. It may also be reasonable to assume that the PA-GFP-TfR signal could be diluted away upon arrival to one compartment to further trace its subsequent movement to the sorting endosome. Indeed, we could not trace the PA-GFP-TfR much longer after its arrival to the Tfn-containing endosomal compartment. Further experiments are necessary to determine definitively whether the recycling endosomes (Ang et al., 2004; Lock JG and Stow JL, 2005; Lock JG et al., 2005) (or other organelles) behave as the intermediates during transport from the Golgi to the plasma membrane and/or from the Golgi to the sorting endosomes.
In conclusion, we have improved the PA-GFP photoactivation technique in terms of accuracy of the extent of photoactivation and harmlessness to live cells, using multiphoton laser. We applied this technique to show that PA-GFP-TfR is delivered to EEA1+ sorting endosomes possibly directly from the TGN. Multiphoton photoactivation monitored by time-lapse imaging could also be employed to study other intracellular protein trafficking pathways, such as the Golgi-to-endoplasmic reticulum and the plasma membrane-to-endosomal/lysosomal compartment, when appropriate combinations of fluorescent marker proteins (organellar and trafficking markers) are designed and generated.
We would like to thank Dr. Roger Y. Tsien for generously providing the reagent; Drs. Koji Hase, Kazuya Kawano, Takaya Murakami, Hiroyuki Takatsu and Atsuko Yoshino for critical reading of the manuscript. This study was supported in part by Grants-in-Aid for Scientific Research (HO), Scientific Research in Priority Areas (HO) and Protein 3000 Project (HO) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Naito Foundation (HO).
|