| To whom correspondence should be addressed: Kazuhiro Nagata, Department of Molecular and Cellular Biology, Institute for Frontier Medical Sciences, Kyoto University, 53 Shogoin Kawahara-cho, Sakyo-ku, Kyoto 606-8397, Japan. Tel: +81–75–751–3848, Fax: +81–75–751–4645 E-mail: nagata@frontier.kyoto-u.ac.jp Abbreviations: AMS, 4-acetamide-4'-maleimidylstilbene-2,2'-disulfonic acid; ARMET, arginine rich, mutated in early stage of tumors; ATF6, activating transcription factor 6; DTNB, 5,5'-dithiobis(2-nitrobenzoic acid); ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; ERSE, ER stress response element; Grp78, 78-kDa glucose regulated protein; HERP, homocysteine-induced endoplasmic reticulum protein; Hsp70, 70-kDa heat shock protein; MALDI-TOF, matrix assisted laser desorption ionization-time of flight; NF-Y, nuclear factor Y; PBS, phosphate-buffered saline; PDI, protein disulfide isomerase; XBP1, X-box binding protein 1. |
Accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER) stimulates pathways leading to mediation of protein folding by molecular chaperones or degradation by ER-associated protein degradation (ERAD) system (Mori, 2000; Urano et al., 2000; Patil and Walter, 2001; Kaufman et al., 2002). Transcription factors XBP1 and ATF6 play key roles in the unfolded protein response by inducing ER chaperones and ERAD machineries. These transcription factors recognize the ER stress response element (ERSE; consensus sequence CCAATN9CCACG) in the presence of NF-Y (Yoshida et al., 1998, 2000, 2001). ERSEs are frequently found in the promoters of ER chaperone genes, including that of Grp78, an Hsp70 family protein required for protein quality control in the ER.
A second ER stress response element, ERSE-II (ATTGGNCCACGT), was discovered in a promoter assay of the human homocysteine-induced endoplasmic reticulum protein (HERP) gene (Kokame et al., 2000, 2001; Yamamoto et al., 2004). HERP is a ubiquitin-domain protein that localizes in the ER membrane and may be involved in quality control of proteins in the ERAD pathway (Hori et al., 2004; Schulze et al., 2005; Nogalska et al., 2006). ERSE and ERSE-II are known to be differentially recognized by ATF6 and XBP1 (Yamamoto et al., 2004). However, the general role of the ERSE-II element in ER stress-responsive genes is poorly understood due to the lack of other examples of the gene containing ERSE-II elements.
Pancreatic β cells produce a high level of insulin and undergo a high level of ER stress (Iwawaki et al., 2004). The diabetes mellitus mouse strain, Insulin-Akita (Yoshioka et al., 1997), carries a C96Y mutation in the Ins2 gene (Wang et al., 1999). Since Cys96 is essential for a disulfide bond in insulin, proinsulin carrying this mutation becomes structurally unstable and is prone to aggregation (Ron, 2002) due to an exposed hydrophobic surface (Yoshinaga et al., 2005). Previously, we established β cell lines from Ins2+/Akita mice and reported that ER chaperones and the ATF6 and XBP1 transcription factors are more strongly activated in Ins2+/Akita cells than in Ins2+/+ cells (Nozaki et al., 2004).
The arginine rich, mutated in early stage of tumors (ARMET) gene was first reported as a human gene highly mutated in a variety of cancers (Shridhar et al., 1996). In a DNA microarray study, ARMET was upregulated by tunicamycin treatment in wild-type cells but not in XBP1 knockout cells in which ATF6 was knocked down with an RNAi vector (Lee et al., 2003). This suggests that ARMET is an ER stress-responsive gene. Here we report that ARMET is an ER-resident soluble protein containing intramolecular disulfide bonds. ARMET expression is upregulated in pancreatic β cells expressing the C96Y mutant of insulin and is induced by a variety of reagents that cause ER stress. Moreover, we found that the unfolded protein response of the ARMET gene is mediated by an ERSE-II element, like the HERP gene. These results suggest that ARMET is involved in protein quality control in the ER.
Mouse pancreatic β cell lines carrying the Ins2+/+ and Ins2+/Akita genes were cultured as described previously (Nozaki et al., 2004). HeLa, mouse L929, NIH 3T3, and BALB/c 3T3 cells were cultured in Dulbecco’s modified essential medium supplemented with 10% fetal bovine serum.
ARMET cDNA was amplified from a mouse cDNA library (Clontech, Palo Alto, CA) by PCR and cloned into the pCAGGS vector (Niwa et al., 1991). A FLAG tag sequence was inserted in ARMET immediately upstream the C-terminal RTDL sequence using PCR, and the product was cloned pCAGGS. Based on the sequence of the mouse ARMET gene, a 600-bp fragment of the ARMET promoter was amplified by PCR from mouse genomic DNA and cloned into the KpnI-XhoI sites of the pGL3-Basic firefly luciferase vector (Promega, Madison, WI). The ERSE-II sequence on the ARMET promoter was disrupted by site-directed mutagenesis. The effector construct containing mouse wild-type Ins2 and its C96Y mutant was described previously (Nozaki et al., 2004). The reporter gene constructs driven by the human wild-type and ERSE-disrupted GRP78 promoters were described previously (Yoshida et al., 1998). Nucleotide sequences of clones were confirmed by sequencing. Supercoiled plasmid DNA was purified by cesium chloride ultracentrifugation.
Cells were plated in 35-mm dishes, cultured for 24 h, and transfected with a mixture of reporter, effector, and internal control plasmid DNA (1.2–2.0 μg depending upon cell types) using 10 μl of LipofectAMINE reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. After a 5-h exposure to the DNA-LipofectAMINE complex, cells were cultured in medium containing 10% serum for 19 h. The luciferase activities of transfected cells were determined using a dual luciferase assay system (Promega) according to the manufacturer’s instructions, and the activity of firefly luciferase was normalized to that of the sea pansy enzyme.
cDNA probes for mouse ARMET and Grp78 mRNA were generated by PCR. To induce ER stress, cells were incubated with a final concentration of 5 μg/ml tunicamycin, 0.3 μM thapsigargin, or 2 mM DTT. Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA). RNA aliquots (10 μg) were denatured and subjected to electrophoresis in a 1% agarose gel containing 2.2 M formaldehyde, 20 mM MOPS-NaOH (pH 7.0), 5 mM sodium acetate, and 1 mM EDTA. RNA was transferred onto a nylon membrane using standard capillary blotting techniques. A mouse adult tissue blot and embryo-stage blot were purchased from Seegene (Seoul, Korea). These filters were hybridized with 32P-labelled probes in PerfectHyb Hybridization Solution (Toyobo, Tokyo, Japan) at 65°C for 1 h and washed in 0.1×SSC/0.1% SDS at 65°C.
Mouse ARMET cDNA was amplified by PCR using the primers 5'-CATATGCTGCGGCCAGGAGACTGTGAAG-3' and 5'-GGATCCCTACAGATCAGTCCGTGCCGCTGG-3'. The amplified fragment containing the ORF of mature ARMET was subcloned into the NdeI and BamHI sites of pET-21a (Novagen, Madison, WI) modified with a N-terminal 6xHis tag. Escherichia coli Origami B(DE3) cells (Novagen) carrying the ARMET expression plasmid were grown at 37°C until the OD600 reached 0.6, at which time the expression of the recombinant protein was induced by addition of 0.4 mM isopropyl 1-thio-β-D-galactoside. After further culture overnight, the cells were collected and sonicated in 20 mM Tris-HCl (pH 8.0) containing 50 mM NaCl. The lysate was centrifuged (16,000×g, 30 min, 4°C). The supernatant was applied to a Super Q Toyopearl column (Tosoh, Tokyo, Japan) equilibrated with 20 mM Tris-HCl (pH 8.0) at 4°C, and the flowthrough fraction was recovered. Further purification was performed at 4°C using the following columns and conditions: HiTrap Heparin (GE Healthcare, Piscataway, NJ, USA) equilibrated with 20 mM Tris-HCl (pH 8.0) and eluted with a linear gradient of 0–1 M NaCl; Bio-Scale CHT2-I (Bio-Rad, Hercules, CA, USA) equilibrated with 10 mM potassium phosphate buffer (pH 7.0) and eluted with a 10–500 mM potassium phosphate gradient; and HiLoad 16/60 Superdex 75 pg (GE Healthcare) equilibrated with 20 mM Tris-HCl (pH 8.0) containing 150 mM NaCl. The purified bovine ARMET was pooled and stored at 4°C.
An antibody against ARMET was produced by immunizing rabbits with a purified recombinant His-ARMET protein. A mouse monoclonal anti-actin antibody (C4) was obtained from Chemicon (Temecula, CA, USA). Cells were lysed in lysis buffer (1% Nonidet P-40, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4) and the supernatant was recovered after centrifugation (12,000×g, 20 min). The protein concentration was determined using the method of Bradford (Bradford, 1976). Proteins (10 μg/μl) were boiled in SDS-PAGE sample buffer containing 0.1 M dithiothreitol and separated by SDS-PAGE. Proteins were transferred onto Immobilon-P membranes (Millipore, Bedford, MA, USA). The membranes were blocked in PBS containing 5% skim milk and 0.05% Tween 20 and incubated with primary antibodies. After incubation with an alkaline phosphatase-conjugated anti-IgG secondary antibody (Biosource, Nivelles, Belgium), the specific binding of antibodies was visualized using tetrazolium bromochloroindolylphosphate/nitrobluetetrazolium (Sigma, St. Louis, MO, USA) solution.
HEK293 cells transfected with FLAG-tagged mouse ARMET were scraped in phosphate-buffered saline (PBS), collected at 100×g for 10 min at 4°C. Cells were suspended in 50 mM Tris-HCl buffer (pH 7.4) containing 5 mM EDTA and kept on ice for 10 min. Immediately after homogenization by passage 20 times through a 0.5-mm gauge needle, the equal volume of 50 mM Tris-HCl buffer (pH 7.4) containing 5 mM EDTA and 0.88 M sucrose was added to avoid osmotic shock. Cell debris and nuclei were removed by centrifugation at 1,000×g for 10 min at 4°C and mitochondria were removed by centrifugation at 12,000×g for 30 min. Microsome fraction was recovered by ultracentrifugation at 100,000×g for 1 h at 4°C. The pelleted microsomes were resuspended in 50 mM Tris-HCl (pH 7.4) containing 5 mM EDTA and 0.44 M sucrose, 0.1 M Na2CO3 (pH 11.3) or 20 mM HEPES-NaOH (pH 7.4) containing 0.15 M NaCl and 1% Nonidet-P40. After ultracentrifugation at 100,000×g for 1 h at 4°C, the resulting supernatants and pellets were subjected to SDS-PAGE followed by immunoblotting using anti-BiP (BD Biosciences, San Jose, CA), anti-calnexin (BD Biosciences), or anti-FLAG® M2 (Sigma, St Louis, MO) monoclonal antibodies.
HEK293 cells transfected with FLAG-tagged mouse ARMET were scraped in PBS. To prevent post-lysis formation of disulfide bonds, trichloroacetic acid (TCA) (final concentration 10%) was immediately added to the cell suspension. The precipitate was dissolved in 0.1 M sodium phosphate (pH 7.0), 1% SDS, and 3 mM 4-acetamide-4'-maleimidylstilbene-2,2'-disulfonic acid (AMS). Free sulfhydryl groups of cysteine residues were modified with AMS by incubation for 20 min at room temperature. FLAG-ARMET proteins used as oxidized and reduced standards were prepared as follows: HEK293 cells transfected with FLAG-tagged mouse ARMET were lysed in 20 mM HEPES-NaOH (pH 7.4) containing 0.15 M NaCl and 1% Nonidet-P40, and supernatant after centrifugation (12,000×g for 30 min at 4°C) was incubated in 10 mM diamide for 20 min at room temperature or boiled in 10 mM dithiothreitol (DTT) for 5 min. After TCA precipitation and modification with AMS, samples were subjected to 15% SDS-PAGE in a reducing condition and followed by immunoblotting with the anti-FLAG® M2 antibody.
All of the following procedures were performed at 4°C. Bovine testis was homogenized in homogenization buffer (50 mM HEPES/KOH [pH 7.4], 250 mM sucrose), and the homogenate was centrifuged at 16,000×g for 30 min and subsequently at 180,000×g for 1 h. The supernatant was subjected to column chromatography as described for the purification of recombinant ARMET. In each step of the purification, fractions containing ARMET were identified by western blotting. Purified ARMET was resolved by SDS-PAGE (15% acrylamide gel) and blotted onto a polyvinylidene difluoride membrane, and its N-terminal amino acid sequence was determined by a protein sequencer.
Chemical modification of the free sulfhydryl groups of proteins was performed in 0.1 M MOPS buffer (pH 7.0) at 37°C in the presence of 0.2 mM 5,5'-dithiobis(2-nitrobenzoic acid) (DTNB), and the reaction was monitored at 412 nm. The number of modified cysteines per protein was determined using a molar extinction coefficient of the resultant product, nitrothiobenzonate (ε412=14100 M–1 cm–1).
Purified recombinant ARMET was cleaved by lysyl endopeptidase, and the molecular mass of resulting fragments was determined by matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry using a Voyager-DE STR instrument (Applied Biosystems, Foster City, CA).
BALB/c 3T3 cells were washed in PBS and fixed in 4% paraformaldehyde for 15 min. Cells were permeabilized by 0.2% Triton X-100 for 4 min, followed by an incubation in 5% normal goat serum and 5% BSA for 30 min. Cells were incubated with a rabbit anti-ARMET antibody and a mouse antibody against protein disulphide isomerase (PDI), an ER-resident marker protein (Stressgen, Victoria, Canada) for 1 h, and then with an Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen). After washes with PBS, localization of fluorescent signals was analyzed using a 510 META confocal laser-scanning microscope (Carl Zeiss, Jena, Germany).
Sections of mouse tissues were immunostained using the avidin-biotin complex method with alkaline phosphatase or peroxidase as described previously (Toyokuni et al., 1997).
We previously established pancreatic β cell lines carrying the Ins2+/+ or Ins2+/Akita genes and found that the latter underwent continuous ER stress (Nozaki et al., 2004) because of misfolding of proinsulin due to a C96Y mutation (Yoshinaga et al., 2005). During the course of an oligonucleotide-based gene array analysis comparing mRNA expression levels between the β cell lines derived from Akita (Ins2+/Akita) and wild-type mice, we found that the expression level of ARMET mRNA was significantly higher in Ins2+/Akita cells than in Ins2+/+ cells (data not shown). A Northern blot analysis confirmed the higher ARMET mRNA level in Ins2+/Akita (Fig. 1A); ARMET mRNA expression was also induced by tunicamycin, an N-glycosylation inhibitor known to induce ER stress in Ins2+/+ cells (Fig. 1A). We also tested the effect of two additional ER stress inducers, thapsigargin (ER-resident Ca2+-ATPase inhibitor) and DTT (reducing agent), using mouse NIH 3T3 fibroblast cells and found that these stress inducers also stimulate the expression of ARMET mRNA (Fig. 1B). The time course of ARMET mRNA induction by thapsigargin or DTT resembles that of Grp78 (Fig. 1C), suggesting that ER stress may induce these two genes by similar mechanisms. The anti-ARMET antibody detected an 18-kDa band in a Western blot analysis, and the band intensity increased after ER stress treatment (Fig. 1D), confirming that the induction also occurs at the protein level. Thus, ARMET is a protein whose expression is stimulated by ER stress. The amount of ARMET expression appears to be mainly determined at the mRNA level, although these observations do not exclude the possibility of further regulation at the protein level.
 View Details | Fig. 1. Expression of the mouse homologue of ARMET is induced by a variety of ER stressors. (A) mRNA expression levels of ARMET in mouse Ins2+/+ (Wild) and Ins2+/Akita (Akita) pancreatic β cells. Total RNA was isolated from Ins2+/+ β cells treated with 5 μg/ml tunicamycin for 4 h, untreated Ins2+/+ β cells as a control, or Ins2+/Akita β cells that express C96Y mutant insulin. Total RNA (10 μg/lane) was hybridized with 32P-labeled ARMET or Grp78 probes. Ethidium bromide staining of total RNA is shown at the bottom. (B) Total RNA from NIH 3T3 cells treated with 0.3 μM thapsigargin, 5 mM DTT, 5 μg/ml tunicamycin for 4 h, or left untreated as a control, were analyzed by Northern blotting. (C) NIH 3T3 cells were treated with thapsigargin or DTT for the indicated periods and analyzed by Northern blotting. (D) Western blot analysis of mouse ARMET. Total cell lysates from NIH 3T3 cells treated with tunicamycin were subjected to Western blot analysis using anti-ARMET, anti-Grp78, and anti-actin antibodies. The ARMET antibody detected an 18-kDa band whose intensity was increased by ER stress. |
The distribution of ARMET mRNA in mouse tissues was partly similar to that of Grp78 mRNA, although the patterns were distinct in several tissues (Fig. 2A). For example, expression levels of these mRNAs are high in the muscle and testis relative to those in the liver, spleen, kidney, stomach, intestine, thymus and uterus. However, the levels significantly differ in the brain, heart, lung and placenta. These results suggest that regulation of these two genes is similar in part, but not identical, in vivo. Because we isolated ARMET as a gene upregulated in cultured pancreatic β cells of the Akita mouse, we further analyzed pancreatic tissues of wild-type and Akita mice by immunohistochemical staining using an antibody specific to ARMET. Strong staining was observed in the pancreatic islets of both wild-type and Akita mice, and the staining intensity was stronger in the islets of the Akita mouse (Fig. 2B and 2C). The Northern blot analysis (Fig. 2A) also indicated that ARMET mRNA is highly expressed in the testis; we therefore also examined testis tissues by immunostaining. Strong signals were detected in spermatocytes (Fig. 2D and 2E), with an expression pattern that closely resembles that of Grp78 (Huo et al., 2004).
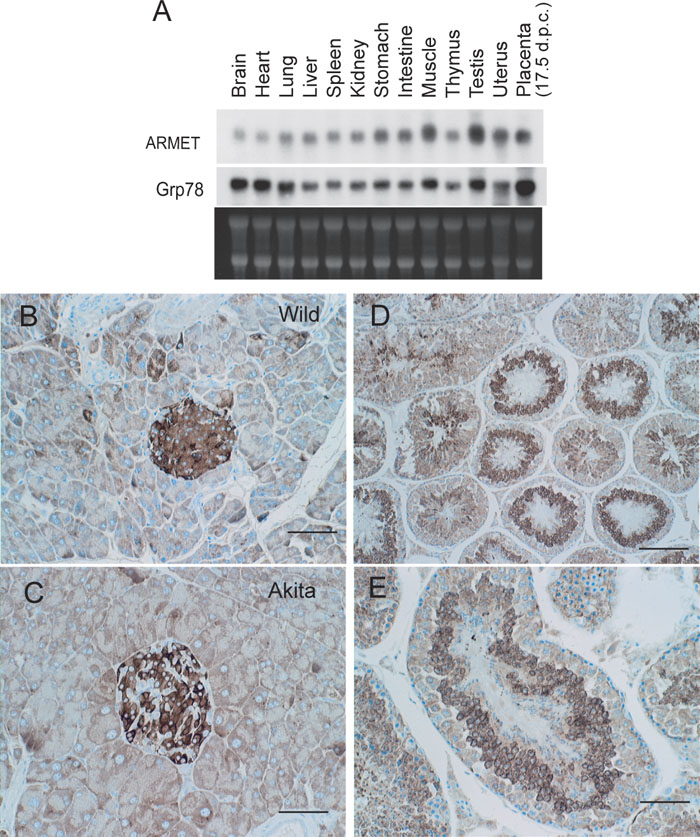 View Details | Fig. 2. Expression patterns of ARMET in mouse tissues. (A) Northern blot analysis of ARMET and Grp78 in adult mouse tissues. (B–E). Immunohistochemical analysis of ARMET. Pancreases from Ins2+/+ (B) and Ins2+/Akita (C) mice and testes from Ins2+/+ mice (D and E) were analyzed by immunostaining using an anti-ARMET antibody. Bar=50 μm for B, C, and E, and bar=100 μm for D. Positive signals are brown, and nuclear staining is blue. |
To determine the subcellular localization of ARMET, we performed immunofluorescent staining of cultured cells using an anti-ARMET antibody and an anti-PDI antibody. The staining patterns of ARMET and PDI overlapped extensively, indicating that ARMET is an ER-resident protein (Fig. 3A). We further analyzed ARMET localization in the ER by alkaline extraction from microsomes using cells expressing FLAG-tagged ARMET. Most FLAG-ARMET protein was extracted by Na2CO3 alkaline treatment whereas calnexin, a typical membrane-integrated protein in the ER, was hardly extracted by this treatment (Fig. 3B). The alkaline extraction of ARMET was even more efficient than that of BiP, a molecular chaperone known to localize in the luminal space of ER and interact with membrane proteins. These results indicate that ARMET is an ER luminal protein that is not integrated to the membrane. Alignment of the amino acid sequences of ARMET homologues from different organisms indicated that ARMET is a highly conserved protein (Fig. 3C). All the ARMET homologues have KDEL-like sequences at the C-terminus, which are known to function as ER-retention signals; this is consistent with the ER localization of ARMET we observed with immunostaining. Bovine ARMET purified from testis (Fig. 3D) had the same mobility as the mouse ARMET detected by Western blotting (Fig. 1D), and peptide sequencing of bovine ARMET indicated that the N-terminal sequence of endogenous ARMET is LRQGDCEVCI (see Fig. 3C). Thus, the polypeptide composed of the first 21 amino acids is cleaved off during maturation. As these 21 residues are hydrophobic and typical of membrane-inserted regions, we consider this to be a signal peptide removed from the ARMET precursor during translocation into the ER.
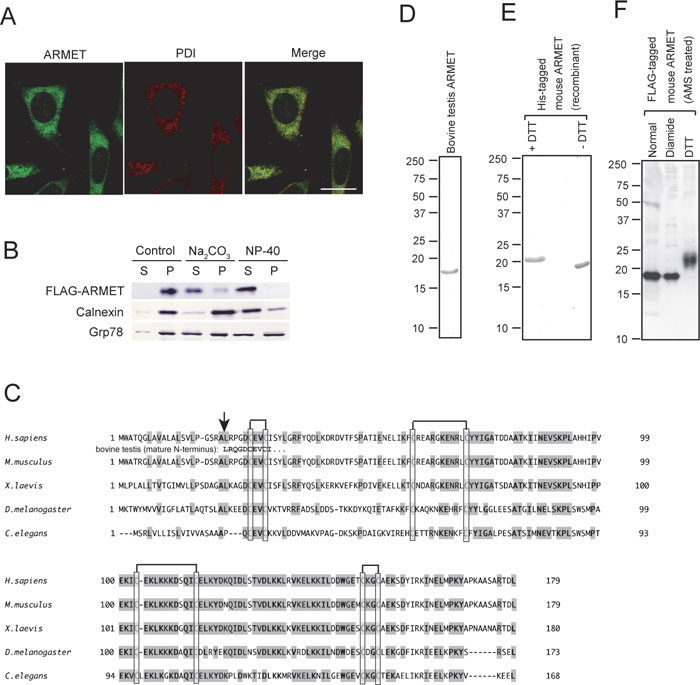 View Details | Fig. 3. ARMET is a soluble protein localized in the ER. (A) Immunofluorescent analysis of Balb/c 3T3 cells. Cells were doubly stained with a rabbit anti-ARMET antibody followed by an Alexa 546-conjugated anti-rabbit IgG and a mouse anti-PDI antibody followed by an Alexa 488-conjugated anti-mouse IgG. ARMET and PDI are shown in green and red, respectively, using pseudocolor. Bar=20 μm. (B) Alkaline extraction of microsomal ARMET. Microsome fraction prepared from HEK293 cells transiently expressing FLAG-ARMET was further fractionated by centrifugation after Na2CO3 or NP-40 treatments or no treatment as a control. Supernatant (S) and pellet (P) fractions were analyzed by western blotting using antibodies against FLAG, calnexin or Grp78. (C) Alignment of amino-acid sequences of ARMET homologues. Amino acid sequences of human, mouse, frog, fruit fly, and nematode ARMET were deduced from cDNA sequences. Completely conserved residues are indicated by bold characters. Cysteine residues are boxed and the pairs forming disulfide bonds are shown by lines at the top. Highly conserved residues are indicated by shading. The N-terminal sequence of bovine ARMET determined by amino acid sequencing of purified protein is shown between the human and mouse sequences. The arrow indicates the signal peptide cleavage sites. (D and E) SDS-PAGE analysis of ARMET purified from bovine testis (D) and His-tagged mouse ARMET purified from E. coli carrying a recombinant plasmid vector (E). (F) All eight cysteine residues of ARMET form disulfide bonds in vivo. FLAG-ARMET was expressed in HEK293 cells. Cell lysates were immediately precipitated by TCA to avoid artificial post-lysis disulfide bond formation. After free cysteine residues were modified by AMS, FLAG-ARMET was analyzed by western blotting using anti-FLAG antibody. To produce oxidizing and reducing conditions, diamide and DTT were added to cell lysates, respectively. |
All of the ARMET homologues have eight conserved cysteines, and four of the eight residues form two CXXC motifs. We also purified His-tagged mouse ARMET from E. coli using a recombinant expression system. The mouse ARMET migrated more quickly in SDS-PAGE gels in non-reducing conditions (Fig. 3E, right) than in reduced conditions (Fig. 3E, left), indicating that the molecule is smaller in non-reducing conditions. In gel filtration chromatography, both endogenous and recombinant ARMET proteins migrated to a position consistent with that of a monomer (data not shown). DTNB titration of the recombinant ARMET revealed that there are no free sulfhydryl groups (data not shown). Thus, the eight cysteine residues form intramolecular disulfide bonds. We further analyzed the location of the disulfide bonds by MALDI-TOF mass spectrometry and found polypeptides containing disulfide bonds at Cys7-Cys10, Cys41-Cys52, and Cys128-Cys131 (numbered from the N-terminus of mature ARMET; data not shown). As ARMET has no free sulfhydryl groups, the remaining cysteines at positions 83 and 94 appear to form disulfide bonds, although this was not detected by mass spectrometry probably because this peptide is highly susceptible to lysyl endopeptidase digestion due to the abundance of lysine residues between Cys83 and Cys94. Thus, ARMET likely has 4 intramolecular disulfide bonds (see Fig. 3C). The formation of disulfide bonds in ARMET in vivo was further confirmed by the lack of free cysteine residue modifiable by AMS. FLAG-tagged ARMET was expressed in cultured cells and directly precipitated by TCA to avoid artificial disulfide bond formation. After free cysteine residues were modified by AMS, AMS modification on FLAG-ARMET was analyzed by immunoblotting under reducing conditions. FLAG-ARMET migrated as an 19 kDa band under diamide oxidizing conditions or normal untreated conditions (Fig. 3F). Since this mobility is identical to that of non-modified FLAG-ARMET (data not shown), these results indicate that ARMET has no free sulfhydryl groups under normal conditions in vivo. In contrast, FLAG-ARMET modified by AMS after DTT treatment migrated as a 23 kDa band (Fig. 3F). Since the molecular weight of AMS is approximately 0.5 kDa and the shift up observed after DTT treatment was 4 kDa, all 4 disulfide bonds produced in ARMET in vivo were cleaved into cysteine residues by DTT and modified with AMS. Another highly conserved characteristic of ARMET homologues is an abundance of charged residues. Consistent with this observation, ARMET is highly soluble even at very high concentrations (data not shown). These observations indicate that ARMET is an ER-resident soluble protein containing intramolecular disulfide bonds.
To analyze the mechanisms by which ER stress induces ARMET gene expression, we produced a luciferase reporter construct driven by the mouse ARMET promoter. We previously found that the C96Y mutant of Ins2 protein stimulates ERSE-dependent transcription from the GRP78 promoter in pancreatic β cells (Nozaki et al., 2004). Here, we overexpressed the C96Y mutant protein in wild-type β cells by transient transfection in the presence of the ARMET-luciferase reporter construct; the wild-type protein was used as a control. The mutant Ins2 induced substantially higher ARMET promoter activity than did the wild-type Ins2 (Fig. 4A). In contrast, no difference was observed with the control reporter construct lacking the ARMET promoter. To test the effect of ER stress on ARMET promoter-dependent expression, NIH 3T3 cells were transfected with a reporter construct driven by the ARMET promoter in the presence and absence of thapsigargin. Following a 6-h treatment with thapsigargin, luciferase activity increased 2.5-fold over the activity detected under normal conditions (Fig. 4B). Similar results were obtained using a construct driven by the wild-type GRP78 promoter, whereas the ERSE-disrupted construct did not respond to the ER stress. These results suggest that the ARMET promoter has ERSE-like activity induced by ER stress. Consistent with this, a reporter gene containing the ARMET promoter (600 bp) was induced 14-fold by tunicamycin treatment in HeLa cells, and deletion of 300 bp from the 5' end had no significant effect on the activity under either normal or stress conditions (Fig. 4E).
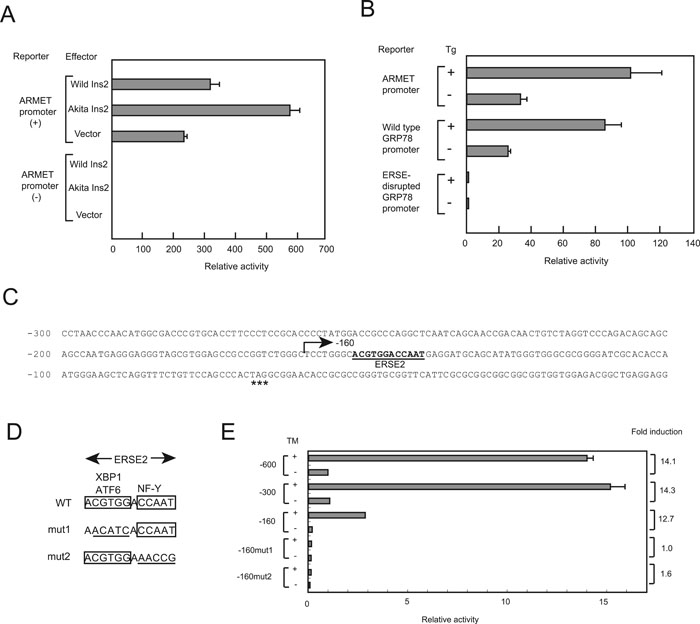 View Details | Fig. 4. ERSE-II-dependent stress response of the mouse ARMET promoter. (A) The C96Y mutant of Ins2 stimulates transcription from the ARMET promoter in pancreatic β cells. Wild-type β cells were co-transfected with a firefly luciferase reporter construct driven by the ARMET promoter or a construct with no insert as a control (0.55 μg) and an effector plasmid expressing the wild-type Ins2 or the C96Y mutant (0.55 μg), and a pRL-SV40 internal control vector expressing sea pansy luciferase (0.1 μg). The activity of both luciferases was determined for each cell extract, and the activity of the firefly luciferase was normalized to that of the sea pansy enzyme (mean and standard deviation of three experiments). The activity of the construct lacking the ARMET promoter in the absence of exogenously expressed Ins2 is set to 1.0. (B) ER stress stimulates transcription from the ARMET promoter in NIH 3T3 cells. Cells were co-transfected with the reporter construct driven by the ARMET promoter or wild-type or ERSE-disrupted human GRP78 promoter (1.1 μg) and an internal control (0.1 μg). Transcriptional activity of the promoters was analyzed in the presence or absence of thapsigargin treatment (Tg). The activity of the ERSE-disrupted GRP78 promoter in the absence of thapsigargin treatment was set to 1.0. (C) The nucleotide sequence of the mouse ARMET promoter. Nucleotide positions are indicated by the distance from the translation initiation codon ATG. The ERSE-II element is indicated by bold characters, and a stop codon (TAG) located 66 bp upstream from the ATG start codon is indicated by asterisks. (D) Nucleotide sequences of ERSE-II mutants. Mutated residues are underlined. (E) Reporter assay of the mouse ARMET promoter. Mouse ARMET promoter elements extending from –1 to –600, –300, or –160 were subcloned into a luciferase reporter vector, and the mutations shown in panel D were introduced into the –160 promoter. The activities of these constructs were analyzed with a dual-luciferase assay using HeLa cells cultured in the presence or absence of tunicamycin (TM). The activity of the –600 promoter in the absence of tunicamycin treatment was set to 1.0. |
Immediately downstream of –160 bp in the ARMET promoter there is an element whose sequence (ACGTGGNCCAAT) completely matches that of the ERSE-II element found in the HERP promoter (Kokame et al., 2001). ERSE-II contains two transcription factor recognition sequences: ACGTGG is recognized by ATF6 or XBP1, whereas CCAAT is recognized by NF-Y. Consistent with these observations, deletion of the promoter from –300 to –160 bp had no significant effect on stress inducibility, while activity under normal conditions was greatly decreased (Fig. 4E). The region between –300 and –160 appears to contain cis-acting element(s) that enhance ARMET promoter activity under normal conditions. To analyze the role of the ERSE-II-like element of the ARMET promoter in stress-induced transcriptional activity, we used nucleotide substitutions to disrupt the binding sequences of ATF6/XBP1 or NF-Y in the –160 promoter (Fig. 4D). The stress inducibility of the ARMET promoter was almost completely abolished by these mutations (Fig. 4E), indicating that both ATF6/XBP1 and NF-Y binding sites are required for the ER stress inducibility. Thus, the ERSE-II element found downstream of –160 bp in the ARMET promoter is essential for the induction of transcription by ER stress.
We have shown in this study that the mouse homologue of ARMET is an 18-kDa, ER-resident soluble protein induced by ER stress (Figs. 1–3). ARMET homologues are highly conserved from nematodes to humans, suggesting that this protein performs an indispensable function. The expression of ARMET is induced by the misfolded C96Y mutant of insulin and by agents that cause unfolded proteins to accumulate. Its expression patterns in mouse tissues are similar to those of Grp78, a molecular chaperone that plays a crucial role in the folding of proteins in the ER. The expression pattern is also similar to that of an ERSE-driven GFP reporter in mice (Iwawaki et al., 2004), suggesting that ARMET expression is probably also induced by ER stress in vivo. These observations suggest that ARMET function may be related to cellular events activated under ER stress conditions.
ARMET has two CXXC sequences that are conserved in all ARMET homologues, and these regions have internal disulfide bonds. Although CXXC is a motif frequently found in oxidoreductases of the thioredoxin family (Ellgaard and Ruddock, 2005), ARMET has no oxidoreductase activity as far as we tested by general methods (data not shown). This appears to be consistent with the lack of homology between ARMET and any other known proteins, including those of the thioredoxin family. The CXXC motif is also frequently found in metal binding proteins, including zinc binding proteins (see for example (Banci et al., 2006)). However, atomic absorption spectroscopy did not detect metal ions in our purified recombinant ARMET protein (data not shown). Thus, the exact function of the ARMET protein in the ER remains to be investigated.
A detailed promoter analysis of the mouse ARMET gene indicated that the unfolded protein response of this gene is mediated by the ERSE-II element (Fig. 4). These results are consistent with the previous observations that ARMET mRNA induction by tunicamycin is diminished by RNAi knockdown of ATF6 in XBP1 knockout cells (Lee et al., 2003). The ERSE-II element was first identified in the promoter of the human HERP gene (Kokame et al., 2001), and the gene encodes a ubiquitin-domain ER membrane protein (Kokame et al., 2000). ARMET is the second fully characterized gene known to be regulated by ERSE-II, and our findings thus extend the role of the ERSE-II element to a different type of ER stress-responsive gene.
ERSE and ERSE-II are known to be differentially recognized by ATF6 and XBP1 (Yamamoto et al., 2004), suggesting that ERSE-II differs from ERSE in the stress response. The expression pattern of ARMET is similar in part, but not identical, to that of Grp78, an ERSE-driven chaperone gene. The difference between ERSE and ERSE-II may be responsible for the expression patterns of Grp78 and ARMET, although the contribution of other cis-acting elements or mRNA stability cannot be excluded. As luman/CREB3 was reported to regulate the HERP gene expression in response to ER stress via the ERSE-II element (Liang et al., 2006), this transcription factor may also be involved in ARMET gene expression.
Recent analyses of HERP suggest that this protein functions in the ERAD pathway (Hori et al., 2004; Schulze et al., 2005). An ERSE-II-like element in the promoter region of the human selenoprotein S gene (also called VIMP) is suggested to be responsible for ER stress inducibility, and this gene encodes a component of an ERAD complex (Schulze et al., 2005). Taken together, our observations suggest that ARMET plays a role in protein quality control during ER stress, including the degradation of misfolded proteins.
We thank Mana Kadota for technical assistance, and Akira Kitamura and Yoshihito Ishida for helpful suggestions on immunofluorescence experiments. We also thank Prof. Kazutoshi Mori and Dr. Hidero Yoshida for the reporter gene constructs driven by wild-type and ERSE-disrupted GRP78 promoters and helpful comments to the manuscript.
|