| To whom correspondence should be addressed: Yasuo Takahashi, Cell-based Analysis team, Advanced Core Technology Department, Research and Development Division, Olympus Corporation, 2-3 Kuboyama-cho, Hachioji-shi, Tokyo, 192-8512, Japan. Tel: +81–42–691–7392, Fax: +81–42–691–7396 E-mail: yas_takahashi@ot.olympus.co.jp Abbreviations: FCCS, fluorescence cross-correlation spectroscopy; AOTF, acousto-optic tunable filter; EGFP, enhanced green fluorescence protein; mRFP, monomeric red fluorescence protein; G-D-R, EGFP-DEVD-mRFP2; AOM, acousto-optic modulator; FRET, fluorescence resonance energy transfer; LSM, laser scanning microscopy; APD, avalanche photo diode. |
FCCS is a highly sensitive measurement technique for investigating the interactions of molecules. This method can detect the coincidence of two molecules labeled with fluorophores of different colors in a small detection area on which two laser beams are focused. FCCS has been applied to studies of molecular interactions in homogeneous solutions (Kettling et al., 1998; Doi et al., 2002; Collini et al., 2005; Swift et al., 2006) and in living cells (Saito et al., 2004; Baudendistel et al., 2005; Kim et al., 2005). Unique features of FCCS technology have recently been reviewed by Bacia et al. (2006) with emphasis on its application to living cells.
The conventional FCCS system employs two lasers of different wavelengths and two detection channels to collect their emission signals. The most common pair of fluorescent proteins for dual-color imaging consists of enhanced green fluorescent protein (EGFP) and monomeric red fluorescent protein (mRFP) (Saito et al., 2004). Because of their bright signals, EGFP and mRFP have been preferred as the two fluorophores for FCCS. As they show broad excitation and emission spectra with modest Stokes shifts, however, it is extremely hard to separate the EGFP and mRFP emission signals by simultaneous excitation (Fig. 1). The use of narrow bandpass filters for minimizing emission cross-talk often results in weak signals. Thus, complex mathematical computations are usually required to compensate for cross-talk and fluorescence resonance energy transfer (FRET).
 View Details | Fig. 1. Emission spectra of EGFP and mRFP (modified from the home page of Dr. A. Miyawaki, RIKEN, Japan). In our experiment, EGFP and mRFP were excited at 488 nm and 543 nm (dotted vertical lines), respectively. Detection channels were set at 510–530 nm (Ch1) for EGFP, and the long path above 560 nm (Ch2) for mRFP. Shadowed area in EGFP emission spectra indicates the unexpected leakage of fluorescence in Ch2, called spectral cross-talk. |
Problems of spectral cross-talk can be excluded by several approaches. Conventional FCCS measurement needs two excitations with different wavelengths and two channels, one for each emission signal. In this case it is hard to eliminate cross-talk signals at the second channel for the fluorophore that has a longer wavelength. Recent modern FCCS methods such as 2 photon-FCCS (2P-FCCS) (Kim et al., 2005; Swift et al., 2006) and single wavelength excitation FCCS (SW-FCCS) (Liu et al., 2007) require only one wavelength excitation system. These methods have the advantages of simple equipment and perfect matching of the confocal volume for two distinct fluorophore excitations. However, in these methods, it is difficult to adjust the strengths of excitation power for each fluorophore to diminish the leakage of emission signals of the shorter wavelength fluorophore into the second channel by reducing the strength of the laser power for it. Thus, the problem of cross-talk might not be completely eliminated. For this reason, several approaches have been proposed for segregating the two emission signals by switching laser excitation. For example, the use of an acousto-optic modulator (AOM) to make alternate laser beams has proved to be effective. Müller and colleagues (2005) used pulsed interleaved excitation to achieve reliable measurement of FCCS, multicolor fluorescence imaging and FRET. Thews et al. (2005) also reported successful elimination of cross-talk signals in their FCCS measurements using live cells. However, unlike these techniques using extra modulators for laser pulse generation, a simpler system that can change excitation power at each wavelength is needed.
Therefore, we herein present a unique, convenient method to remove spectral cross-talk for FCCS measurement. The main concept of our system is to make precise alternating laser beams by switching acousto-optic tunable filters (AOTFs) in the excitation laser unit. In our switching FCCS method, we can adjust excitation power to be suitable at each wavelength and completely segregate the cross-talk in each channel. Then synchronized fluorescence signals at detectors are calculated as cross-correlation functions. To demonstrate the reliability of our system, caspase-3 reactions were detected in vitro and in situ through cell apoptosis.
A schematic of switching FCCS is shown in Fig. 2. In conventional FCCS measurement, two lasers simultaneously illuminate samples and the emission signals in their overlapping focal areas are introduced to two distinct APD detectors. Contrary to the previous system, samples are alternately illuminated by each laser via control of the AOTF in our switching FCCS system. As a result, each detector can obtain the expected emission signals in a clear, separate mode. Cross-talk problems are generally caused by signals of green fluorescence leaking into the red channel detector. Theoretically, extraneous signals in the red channel detector are eliminated when alternating laser excitation is carried out by switching. Any combination of the two lasers can be adapted to the two selected fluorophores depending on the purpose of the measurement. In this report, we used a combination of 488 nm and 543 nm excitation for EGFP and mRFP, respectively.
 View Details | Fig. 2. A, B: Schematic of switching FCCS setup. Excitation by two lasers was generated using an AOTF to distribute alternating laser beam exposure of the sample. A: Example of laser 1 excitation. The excitation beam, shown as a blue line, illuminates the sample. The fluorescent light from the sample is then detected by detector 1 in synchronization with laser 1. B: Example of laser 2 excitation. In the same way as for case A, the fluorescent light generated by excitation from laser 2 is detected by detector 2. Switching was done by the switching signal generator at a frequency of 50 kHz. C: The pattern of switching signal processing and detection signals at each APD. (a): AOTF signals controlled with 50 kHz frequency. (b): Rectangular pulses of excitation outputs of lasers. (c): Detection signals at each APD. |
The basic combination of a laser scanning microscope (FV1000, Olympus, Tokyo) and FCCS module was set up as shown in Fig. 2A, B (Takahashi et al., 2005). The signal processing for switching was performed as shown in Fig. 2C. For switching excitatory laser beams, we set the AOTF to obtain alternating laser wavelengths of 488 nm and 543 nm for EGFP and mRFP, respectively. The frequency of switching excitation laser beams was set to 50 kHz. The excitation beams were focused via a water immersion objective lens (UApo, 40X, 1.15 NA; Olympus, Tokyo). The emission signals were divided into two APD channels, a bandpass filter (510–530 nm) for EGFP and a longpass filter (>560 nm) for mRFP. In both in vitro and in vivo experiments, FCCS was performed for 32 sec for each sample, and auto- and cross-correlation functions were obtained.
The autocorrelation functions of the red, Gr(τ) and green fluorescence, Gg(τ), and also the cross-correlation functions, Gc(τ) were calculated by the following equation (1).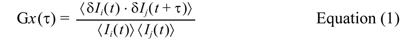
where Ii is the florescence intensity of the green channel (i=g) or the red channel ( j=r), τ denotes the time delay, and Gx(τ) denotes the auto-correlation functions (i=j=x=g or i=j=x=r), and cross-correlation (i=g, j=r and x=c), respectively.
Obtained auto- and cross-correlation functions were fitted to the model as follows: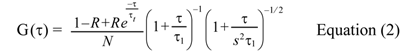
where τ1 denotes the diffusion time of the fluorophore, s=wz /wxy denotes the axial ratio of the volume element, and N denotes the number of fluorescent molecules in the confocal volume with the radius wxy and length 2wz, while R denotes the triplet state fraction, and τt denotes the triplet state transition time. This fitting analysis was performed using the Levenberg-Marquard algorithm of OriginPro 7.5J (OriginLab Corp., Northampton, MA, USA). Quantitative analysis of cross-correlation was done as in previous reports (Kogre et al., 2006; Saito et al., 2004). Relative cross-correlation amplitude was calculated as normalization of the cross-correlation amplitude, Gc(0) by the GFP auto-correlation amplitude, Gg(0). The relative cross-correlation amplitude was written as Gc(0)/Gg(0).
HeLa cells were grown in a 5% CO2 humidified atmosphere at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum. They were plated on LAB-TEK chambered coverslips (Nalge Nunc International, Naperville, IL, USA), and DMEM was changed to phenol red-free medium (OPTI-MEM; Gibco, Japan) before FCCS measurement.
Caspase-3 recognition sites (DEVD) were engineered in the N-terminus of the tandem mRFP dimer (mRFP2) and inserted into the plasmid pEGFP-Cl (Clontech) as previously described by Saito et al., 2004). Fluorescent proteins (EGFP, mRFP2, and EGFP-DEVD-mRFP2; G-D-R) were produced and purified by a previously reported protocol (Saito et al., 2004). These plasmids were transfected into HeLa cells with Effectene (Qiagen, Germany) following the manufacturer’s instructions.
For in vitro assay, chimera proteins (G-D-R) were incubated with caspase-3 (0.07 μg/μl) for one hour at room temperature. FCCS measurement was performed in buffer and cross-correlation functions were calculated. The time-course changes of the amplitude (Gc(0)) decreases due to enzyme cleavage of proteins with and without switching were compared. Proteolysis assay was carried out using HeLa cells transfected with cDNA coding for G-D-R, and the cells were treated with 0.5 μg/ml anti-Fas apoptosis-inducing antibodies (Sigma, USA), and 0.5 μg/ml cyclohexamide (Sigma). After 4 hours of treatment, we confirmed that most cell shapes were completely changed. As a negative control, cells were transfected with a mixture of equal amounts of plasmids coding for EGFP and mRFP2. We compared how much intrinsic caspase-3 in apoptotic cells cleaved the fluorophore substrate G-D-R by FCCS with and without switching.
Fig. 2A & B show the mechanisms of the switching FCCS system. Samples on the microscope stage were exposed to alternating two laser beams (indicated as Laser 1 [blue] and Laser 2 [red] in Fig. 2). The fluorescence signals from samples were then detected by two detectors (APD1 and APD2 for the excitations by Laser 1 and Laser 2, respectively). These data were collected and calculated in the data processing unit. The 50 kHz switching mode was generated by the switching signal generator (Fig. 2C). The pattern of laser excitation is a rectangular pulse as indicated in Fig. 2C(c). To get stable excitation of samples from each laser, a suitable exposure duration was required. For this reason, we chose 50 kHz as a switching frequency. Lower frequencies such as 25 kHz and 12.5 kHz were no different from 50 kHz on autocorrelation curves and fitting results (data not shown).
We carried out a simple experiment to evaluate if our new switching system could eliminate leakage of green fluorescence into the red channel (cross-talk) using EGFP. To mimic conventional FCCS measurement for two fluorophores, the EGFP in solution was excited by a 488 nm laser beam, and two detection channels were opened to detect the EGFP emission signals. When the switching system was off, fluorescence from EGFP was detected in the red channel and, therefore, pseudo cross-correlation functions were observed. On the other hand, this pseudo cross-correlation disappeared when EGFP was measured by switching FCCS (Fig. 3).
 View Details | Fig. 3. Cross-correlation functions of EGFP measured with and without switching FCCS. Without switching, fluorescence of EGFP leaked into the red channel and significant cross-correlation amplitude was observed (black lines of the measured curve and its fitted curve). When EGFP was measured by switching FCCS, cross-talk was removed and no false cross-correlation was detected (red measured and fitted lines). |
We further tested in vitro FCCS measurement for a mixture of two proteins, EGFP and mRFP2 (G/R mix), in solution. We observed that the value of the relative cross-correlation amplitude of the G/R mix was 0.12 when the switching system was off. This value decreased significantly to 0.04 when the switching system was on. Thus, the new switching FCCS was confirmed to enhance the signal-to-noise ratio of cross-correlation functions by removing spectral cross-talk.
The site of cleavage (Asp-Glu-Val-Asp: DEVD) by caspase-3, which specifically acts in the process of cell apoptosis (Shi, 2002), was inserted between EGFP and mRFP2. Fig. 4 shows the kinetics of the protease reaction measured by FCCS with and without switching. The decrease of relative cross-correlation amplitude due to cleavage by caspase-3 was monitored before and after enzyme treatment in buffer solution. The inset graphs in Fig. 4 denote the cross-correlation functions before and one hour after caspase-3 treatment. When switching was on (right inset graph), the curve became almost flat after one hour (red fitted line in right inset). On the other hand, as shown in the left inset graph, some level of amplitude still remained at one hour after enzyme treatment when switching was off (red fitted line in left inset).
 View Details | Fig. 4. Switching effect on kinetics of caspase-3 cleavage in solution. Time-course analysis of relative cross-correlation amplitudes of G-D-R after injection of caspase-3. The dotted and solid lines show the results obtained by FCCS measurements without and with switching, respectively. Inset graphs represent cross-correlation functions before and one hour after caspase-3 treatment. The left inset graph shows a result for FCCS measurement without switching. The right one shows a result obtained with switching. In both graphs, the black measurement lines and fitted lines indicate cross-correlation functions before enzyme treatment. Red measurement and fitted lines indicate results at one hour after treatment. |
The relative cross-correlation amplitude decreased immediately after caspase-3 treatment, as shown in Fig. 4. The amplitude of relative cross-correlation with switching (Gc(0)/Gg(0)=0.32) was significantly lower than that without switching (Gc(0)/Gg(0)=0.49) before enzyme treatment. When switching was used, the amplitude fell to almost zero (Gc(0)/Gg(0)=0.02) at 60 minutes after enzyme treatment. However, with the switching off, the amplitude of the cross-correlation was still high (Gc(0)/Gg(0)=0.21). The amplitude decrease with switching was much larger (94%) than that of the decrease without switching (57%). We could detect clear changes of enzymatic activity due to a significant decrease of the background signals in FCCS measurement using the switching system.
We further investigated the effectiveness of switching for proteolysis assay in living cells as the activity of caspase-3 increased in the last stage of apoptotic cell death. A plasmid of the chimera protein G-D-R, the same component we used for the in vitro experiment, was transfected into HeLa cells. As a control experiment, two distinct plasmids, EGFP and mRFP2, were cotransfected into cells. Two days after transfection, FCCS measurement was performed, and the effectiveness of the switching was evaluated.
Fig. 5A shows an example of relative cross-correlation functions determined by FCCS measurement in a G-D-R-transfected non-apoptotic cell. In this intact cell, expressed G-D-R proteins were well preserved without cleavage by caspase-3. Only a small difference of relative cross-correlation amplitude between the FCCS measurements with and without switching was observed (red measured and fitted lines). In the case of an apoptotic cell, most G-D-R proteins must be cleaved by activated caspase-3. As we expected, the relative cross-correlation amplitude decreased significantly with switching (Fig. 5B; red measured and fitted lines). On the other hand, some amplitude remained when FCCS was measured without switching (Fig. 5B; black measured and fitted lines). In the negative control, independently moving EGFP and mRFP2 were measured by FCCS. Fig. 5C shows that the false-positive amplitude of relative cross-correlation due to cross-talk was diminished by switching (red measured and fitted lines).
 View Details | Fig. 5. In vivo FCCS measurements of G-D-R or a mixture of EGFP and mRFP2 in HeLa cells. A: Relative cross-correlation functions of G-D-R in non-apoptotic cells. Black measurement and fitted lines represent the FCCS measurement with the switching off. Red fitted lines show results for FCCS measurement with the switching on. B: Relative cross-correlation functions of G-D-R in apoptotic cells. C: Relative cross-correlation functions for the mixture of EGFP and mRFP2. Inset images show the measured cells, and a plus sign (+) represents a measurement point for FCCS. Scale bars, 10 μm. D: Summary of the switching effect on relative cross-correlation amplitude. From left to right: G-D-R in non-apoptotic cells, G-D-R in apoptotic cells, and the EGFP/mRFP2 mixture in non-apoptotic cells. Open squares represent results of FCCS measurements with switching off. Closed squares mean for FCCS with switching on. Error bars represent standard errors of repeated measurements, *: p<0.05, ***: p<0.001 (Student’s t-test). |
Fig. 5D and Table I show the statistical analysis of relative cross-correlation amplitudes (Gc(0)/Gg(0)). In the negative control, EGFP and mRFP2 in cells (G/R mix), we found low amplitude (0.15±0.02; mean±SE) caused by spectral cross-talk when switching was off. By contrasting, with switching, the level of relative amplitude was almost zero (0.01±0.01). When cells were intact without apoptosis-stimulation treatment, their shapes were normal for more than four hours. In this case, the level of caspase-3 activity in cells with cleavage protein G-D-R was low. When the switching was off, the relative cross-correlation amplitude of these intact cells that expressed G-D-R was 0.65±0.04 (mean±SE). In contrast, the value of the relative cross-correlation amplitude of G-D-R in these intact cells measured by switching FCCS was 0.53±0.04. A small difference of amplitude might be caused only by cross-talk. Furthermore, we tested the caspase-3 activity in apoptotic cells. Cell shapes were greatly changed at 4 hours after treatment inducing apoptosis using anti-Fas antibodies and cyclohexamide (inset image in Fig. 5B). In these cells, most chimera proteins might be cleaved by activated caspase-3 due to cell apoptosis. A lower relative cross-correlation amplitude (0.27±0.02) was observed in apoptotic cells by measurement without switching. However, this value for amplitude contained a cross-talk effect and an artifact of false-positive amplitude. Therefore, switching FCCS was performed for the same apoptotic cells, and the relative cross-correlation amplitude was calculated (0.05±0.01). A significant decrease of amplitude was clearly observed when cells were measured with the switching system. This value was slightly higher than that of the control experiment (G/R mix) in intact cells measured by switching FCCS. This difference suggested that caspase-3 could not cleave the entire amount of chimera proteins in apoptotic cells even 4 hours after treatment. In this experiment using apoptotic cells, we confirmed that the kinetics of enzymes in living cells could be evaluated by switching FCCS with a lower background level due to elimination of spectral cross-talk signals.
We developed the switching FCCS system to reduce the false-positive cross-correlation function caused by spectral cross-talk between two fluorophores. As shown in Fig. 2, two alternating laser excitations were controlled by an AOTF and then green and red fluorescence signals were collected in their corresponding green and red channels with a specific frequency. These collected signals were calculated to auto- and cross-correlation functions using a data processing unit.
Since correlation functions are generally calculated using sequential acquisition data of photons, a periodical data deficit caused by switching might be a serious problem. We used a switching frequency of 50 kHz; this means that we continuously lost signals every 20 μsec as indicated in Fig. 2C. To prevent any influence of this missing signal on the calculation of auto- and cross-correlations, we applied different weights to the detected data from each channel depending on the on-off state of switching. We composed weighting tables to present signal segregation, and correlation functions could be calculated using both detection signal data and the weighting table. Correlation functions were obtained using these data, except for the parts where the value was zero due to data loss. The fitting results from the obtained correlation functions were approximately the same without inferiority in comparison with the results measured by the conventional method without switching. Particularly in cross-correlation functions, amplitude of function is an indicator of molecular interaction; therefore, the negative effect of signal loss should be negligible in evaluating the results in this article.
Problems of spectral cross-talk can be excluded by certain approaches. Fine segregation of emission spectra between two fluorophores is a great help to avoid it. Complete segregation of emission signals in each detector by mechanical modification is a promising way to remove cross-talk. This means a setup equipped with opening and shuttering of channels corresponding to the various types of fluorescence. Recently, two methods in which alternating excitation lasers controlled by an AOM (Müller et al., 2005; Thews et al., 2005) were employed for FCCS were reported. Whereas the AOM can only modify a single laser wavelength, our system using an AOTF can control multiple laser beam wavelengths. Since the AOTF can easily change the power output of each laser, various complex conditions depending on the experimental procedure are available. In addition to these advantages, the modern LSM system uses the AOTF; therefore, the switching module can be easily modified.
Using our switching FCCS for proteolysis assay in both in vitro and in vivo FCCS measurements, we could demonstrate that alternating laser excitation worked precisely with modification of the AOTF. A significant decrease of relative cross-correlation amplitude was observed when FCCS measurement was performed in the switching mode. In particular, the activity of caspase-3 in living cells could be precisely monitored with time by using switching FCCS. A high background signal level may be caused by false-positive amplitude, leading to changes in the enzyme reactions being under-evaluated. To judge a quantitative enzyme reaction, we need to remove all background signals. As shown in Fig. 5D, the difference of relative cross-correlation amplitude between G-D-R (+) and G-D-R (–) with switching-on (0.53–0.05=0.48) was more significantly so than that with switching-off (0.65–0.27=0.38). With the elimination of the background signals of cross-correlation amplitude by switching, it would be possible to measure the kinetics of the enzyme more precisely.
FCCS is a powerful technique for detecting molecular interactions with high sensitivity. By adding the switching motif, the biological activity changes in living cells can be evaluated with high reliability. Since the level of cross-talk depends on the difference of emissions by two fluorophores, particularly with the stronger emission power of green fluorescence compared to red, we must pay careful attention to the ratio of the expression levels of the two fluorophore-labeled proteins in living cells. In particular, the overexpression of EGFP in comparison with red proteins may cause a large degree of cross-talk. Using switching FCCS, we can detect all areas of cells without any cross-talk. Therefore, all the molecular interactions occurring in cells can be detected under the same conditions. This is important to understand protein functions and their orchestrated dynamics in living cells.
The authors would like to thank Dr. K. Saito and Dr. Y. Ohsugi (Hokkaido University) for preparation of chimera plasmids and proteins; Dr. T. Sugiyama (Olympus Corporation) for assistance with the in vitro experiment; and Dr. R. Sawada (Olympus Corporation) for development of the FCCS system. This work was supported by grants from NEDO (the New Energy and Industrial Technology Development Organization).
|