| To whom correspondence should be addressed: Department of Biophysics, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwake, Sakyo-ku, Kyoto 606-8502, Japan. Tel: +81–75–753–4067, Fax: +81–75–753–3718 E-mail: hide@biophysics.mbox.media.kyoto-u.ac.jp kazu.mori@bio.mbox.media.kyoto-u.ac.jp Abbreviations: AD, activation domain; b, basic region; DEG, degradation domain; ER, endoplasmic reticulum; ERAD, ER-associated degradation; MEFs, mouse embryonic fibroblasts; NES, nuclear exclusion signal; pre-mRNA, precursor mRNA; UPR, unfolded protein response; ZIP, leucine zipper. |
The accumulation of unfolded proteins in the endoplasmic reticulum (ER) activates a series of translational and transcriptional programs collectively termed the unfolded protein response (UPR). The role of the UPR is to maintain the homeostasis of the ER, where newly synthesized secretory and transmembrane proteins are folded and assembled to gain tertiary and quaternary structures. Signaling from the ER is mediated by transmembrane proteins, which have increased in number with evolution and developed more complex and sophisticated ways of dealing with unfolded protein accumulation (Bernales et al., 2006; Harding et al., 2002; Mori, 2000; Schroder and Kaufman, 2005).
The budding yeast Saccharomyces cerevisiae possesses a single signal transduction pathway (Mori, 2003; Patil and Walter, 2001). ER stress activates Ire1p, a transmembrane protein kinase/endoribonuclease in the ER, resulting in unconventional (frame switch) splicing of HAC1 mRNA, which codes for the yeast UPR-specific transcription factor Hac1p. HAC1 mRNA is constitutively synthesized as an intron-containing precursor mRNA (pre-mRNA) but is not translated because the HAC1 intron, consisting of 252 nucleotides, is capable of blocking translation. Since the 5' splice site is located within the coding region, they share an N-terminal 220 aa region containing a basic leucine zipper (bZIP) domain but differ in their C-terminal tail regions. Thus, HAC1 pre-mRNA encodes Hac1p of 230 aa (unspliced Hac1p) while HAC mature mRNA encodes Hac1p of 238 aa (spliced Hac1p). Interestingly, the C-terminal 18 aa of spliced Hac1p functions as a transcriptional activation domain (AD) whereas the 10 aa of unspliced Hac1p does not. Thus, ER stress-induced splicing of HAC1 pre-mRNA not only allows the synthesis of Hac1p via removal of the intron, but also renders synthesized Hac1p highly active in transcription by joining the DNA-binding domain with the AD (Mori et al., 2000).
Metazoan cells have evolved a second pathway that is initiated by pek-1 or PERK in worm or mammalian cells, respectively, a transmembrane protein kinase in the ER. ER stress-induced activation of these molecules results in a general attenuation of translation via direct phosphorylation of the α subunit of eukaryotic translation initiation factor 2. This response decreases the burden on the affected ER (Ron, 2002). Paradoxically, translational attenuation induces translation of certain mRNAs, in mammalian cells at least, such as ATF4 mRNA, which encodes a bZIP transcription factor (Harding et al., 2000). This translationally induced ATF4 in turn activates the transcription of genes involved in amino acid metabolism and resistance to oxidative stress (Harding et al., 2003).
A third pathway has emerged in mammalian cells, the ATF6 pathway (Mori, 2003; Schroder and Kaufman, 2005). Two types are expressed, ATF6α and ATF6β, both of which are ubiquitously synthesized as transmembrane proteins in the ER, designated pATF6α(P) and pATF6β(P), respectively, and activated by ER stress-induced proteolysis (Haze et al., 2001; Haze et al., 1999). Caenorhabditis elegans possesses a single ATF6 gene, although worm ATF6 plays little role in transcriptional induction in response to ER stress and has not been shown to be activated by proteolysis (Shen et al., 2005). Upon ER stress, mammalian pATF6α/β(P) are transported from the ER to the Golgi apparatus where they are cleaved by the sequential action of site-1 and site-2 proteases (Ye et al., 2000). Cytosolic fragments of ATF6α/β thus liberated from the Golgi membrane carry all the hallmarks of an active transcription factor. These active forms of ATF6α/β, designated pATF6α(N) and pATF6β(N), respectively, enter the nucleus and activate the transcription of genes for ER-localized molecular chaperones and folding enzymes (ER chaperones) (Okada et al., 2002; Yoshida et al., 2000). Analysis of mouse embryonic fibroblasts (MEFs) deficient in ATF6α or ATF6β revealed that ATF6α but not ATF6β is required for transcriptional induction of not only ER chaperones but also components of the ER-associated degradation (ERAD) machinery (Wu et al., 2007; Yamamoto et al., 2007).
The Ire1p homologues in worm and mammalian cells have been identified as ire-1 and IRE1, respectively (Bernales et al., 2006; Mori, 2003). In contrast, the substrates of these two homologues, which each encode a UPR-specific transcription factor, switch from HAC1 mRNA to xbp-1 mRNA in worm cells and to XBP1 mRNA in mammalian cells. Similar to the case of Hac1p, ER stress-induced splicing causes replacement of the C-terminal region of XBP1 (Calfon et al., 2002; Yoshida et al., 2001). As a result, the bZIP domain joins with the AD in the spliced form of XBP1, designated pXBP1(S), which activates transcription efficiently (Yoshida et al., 2001) (see Fig. 1). ER chaperones and ERAD components are considered to be targets of pXBP1(S) (Lee et al., 2003; Oda et al., 2006; Yoshida et al., 2003). Interestingly, the intron of XBP1 pre-mRNA is 26 nucleotides long, which is too short to block translation. It has been shown that XBP1 pre-mRNA is constitutively expressed, albeit at a low level, and indeed translated to produce the unspliced form of XBP1, designated pXBP1(U), in contrast to the case of HAC1 pre-mRNA (Calfon et al., 2002; Yoshida et al., 2001). However, pXBP1(U) produced at low levels is rapidly degraded by the proteasome (Tirosh et al., 2006; Yoshida et al., 2006), and this degradation appears to be mediated by ubiquitin-dependent and -independent mechanisms (Tirosh et al., 2006). Although pXBP1(S) shares the instability of pXBP1(U), XBP1 mRNA is induced in response to ER stress with a time course similar to that of ER chaperone mRNAs (Yoshida et al., 2000). IRE1-mediated splicing of this induced XBP1 mRNA results in the production of higher levels of pXBP1(S), which escapes from proteasome-mediated degradation and activates transcription in the nucleus (Yoshida et al., 2001).
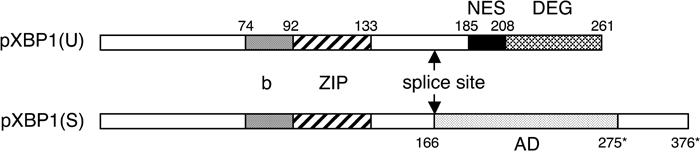 View Details | Fig. 1. Schematic structures of pXBP1(U) and pXBP1(S). Domain structures of pXBP1(U) and pXBP1(S) are schematically shown. ER stress-induced unconventional (frame switch) mRNA splicing replaces the C-terminal region at the position indicated by splice site. b, basic region; ZIP, leucine zipper; NES, nuclear exclusion signal; DEG, degradation domain; AD, activation domain. b functions as a nuclear localization signal and DNA-binding domain. Numbers indicate the C-terminal end of the respective domain. Numbers marked by asterisks denote pXBP1(S)-specific aa. |
We recently found that pXBP1(U) accumulates in the recovery phase of ER stress (Yoshida et al., 2006). That is, although induced XBP1 mRNA is no longer spliced in this phase, translation continues, resulting in the production of high levels of pXBP1(U). pXBP1(S) is localized exclusively in the nucleus, as expected for an active transcription factor, whereas pXBP1(U) is found to shuttle between the nucleus and cytoplasm due to the presence of the nuclear exclusion signal (NES) in the pXBP1(U)-specific C-terminal region (Tirosh et al., 2006; Yoshida et al., 2006) (see Fig. 1). Further, pXBP1(U) forms a complex with pXBP1(S), the pXBP1(U)-pXBP1(S) complex, which is sequestered from the nucleus and degraded by the proteasome due to the presence of the degradation domain (DEG) in the C-terminal end of pXBP1(U) (Tirosh et al., 2006; Yoshida et al., 2006) (see Fig. 1), although the precise role of the DEG in degradation of the pXBP1(U)-pXBP1(S) complex remains to be clarified; one possibility is that it may recruit a specific E3 ubiquitin ligase. This indicates that pXBP1(U) functions as a negative regulator of pXBP1(S). Thanks to this mechanism, the cell can shut off transcription of pXBP1(S)-target genes immediately upon the inactivation of IRE1 that occurs when the ER stress is resolved (Yoshida et al., 2006).
Here, we examined whether pXBP1(U) interacts with and affects other bZIP transcription factors activated in response to ER stress in mammalian cells, specifically ATF6α/β and ATF4.
HeLa cells as well as XBP1+/+ and XBP1–/– MEFs (Lee et al., 2003) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 2 mM glutamine and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin). Cells were maintained at 37°C in a humidified 5% CO2/95% air atmosphere. Transient transfection was carried out by the standard calcium phosphate method (Sambrook et al., 1989) as described previously (Yoshida et al., 1998).
pcDNA-XBP1(U) and its various deletion mutants as well as pCMV-His-XBP1(U) were described previously (Yoshida et al., 2001; Yoshida et al., 2006). ATF4 expression plasmid was the kind gift of Dr. David Ron (New York University). pcDNA-ATF6α(N) and pcDNA-ATF6β(N) were described previously (Haze et al., 2001; Haze et al., 1999).
HeLa cells as well as XBP1+/+ and XBP1–/– MEFs grown in 60 mm dishes were harvested with a rubber policeman and collected by centrifugation. Cell pellets were suspended in 20 μl of ice-cold PBS containing protease inhibitor cocktail (Nacalai Tesque) and 10 μM MG132, mixed with 20 μl of 4× SDS-sample buffer (200 mM Tris/HCl, pH 6.8, containing 400 mM dithiothreitol, 8% SDS, and 40% glycerol), and then boiled for 10 min. Ten-microliter aliquots were subjected to SDS-polyacrylamide gel electrophoresis using 4–20% gradient gels, transferred onto a Hybond-ECL filter (GE Healthcare), and reacted with various antibodies according to the standard protocol (Sambrook et al., 1989), followed by reaction with Western Blotting Luminol Reagent (Santa Cruz Biotechnology). Anti-XBP1-A, anti-ATF6α, and anti-ATF6β antibodies were prepared previously (Haze et al., 2001; Haze et al., 1999; Yoshida et al., 2001). Anti-ATF4 (CREB2) antibody was purchased from Santa Cruz Biotechnology. Chemiluminescence was visualized using an LAS-3000 LuminoImage analyzer (Fuji Film). Protein expression levels were quantified in the range in which the association between chemiluminescence intensity and protein abundance is linear.
HeLa cells grown on cover slips were transiently transfected with appropriate expression plasmid by the calcium phosphate method. Cells were fixed with 2% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and stained with appropriate antibody. Cover slips were mounted with 90% glycerol—10% PBS containing 100 ng/ml of DAPI. Images were acquired using an E800 microscope (Nikon) and ORCA-ER digital CCD camera (Hamamatsu Photonics).
HeLa cells transfected with appropriate expression plasmids were harvested with a rubber policeman, lysed in 600 μl of binding buffer (20 mM HEPES, pH 7.9, containing 100 mM KCl, 10% glycerol, 1 mM MgCl2, 1 mM 2-mercaptoethanol, 0.1% Tween-20, 20 mM imidazole and protease inhibitor cocktail), and then clarified by centrifugation. Supernatants were incubated at 4°C for 1 h with 10 μl of Ni-NTA agarose (Qiagen). The resin was washed with 1 ml of binding buffer three times, suspended in 10 μl of 1xSDS-sample buffer, boiled for 10 min and then centrifuged. Proteins eluted from the resin were subjected to 4–20% SDS-polyacrylamide gel electrophoresis and analyzed by immunoblotting using an appropriate antibody.
To examine the interaction of pXBP1(U) with the cleaved and nuclear form of ATF6 or translationally induced ATF4, HeLa cells were transfected with plasmid to express pATF6α(N), pATF6β(N) or ATF4 together with or without plasmid to express poly-histidine-tagged pXBP1(U) [His-XBP1(U)]. Lysates of transfected cells were incubated with Nickel resin to trap His-XBP1(U), and bound proteins were analyzed by immunoblotting. As shown in Fig. 2, analysis of the eluate clearly showed that pATF6α(N) and pATF6β(N) were retained in the Nickel column via interaction with His-XBP1(U) (compare lanes 10 and 11 with lanes 7 and 8). In contrast, ATF4 was not retained in the Nickel column even when co-expressed with His-XBP1(U) (lanes 9 and 12). These results indicated that pXBP1(U) forms a complex with ATF6α/β but not ATF4.
 View Details | Fig. 2. Association of pXBP1(U) with ATF6 but not ATF4. HeLa cells were transfected with pcDNA-ATF6α(N), pcDNA-ATF6β(N) or pcDNA-ATF4 together with (+) or without (–) pCMV-His-XBP1(U). Lysates of transfected cells were incubated with Ni-NTA agarose. Proteins bound specifically to the resin (eluate) as well as aliquots of lysates [input (10%)] were analyzed by immunoblotting using a mixture of anti-ATF6α, anti-ATF6β, and anti-ATF4 antibodies. |
To clarify the significance of this interaction, we determined the effect of co-expression of pXBP1(U) on the level of pATF6α(N) or ATF4. When HeLa cells were transfected with plasmid to express pATF6α(N) plus vector alone, pATF6α(N) was detected in lysates (Fig. 3A, upper panel, lane 1), and its level was slightly increased by the addition of the proteasome inhibitor MG132 into the culture prior to harvest (lane 6), indicating that MG132 blocked degradation of ATF6 by the proteasome (see quantification data below). Co-expression of pXBP1(U) with pATF6α(N) markedly decreased the level of pATF6α(N) (compare lane 2 with lane 1), but the addition of MG132 restored the level of pATF6α(N) in cells co-expressing pXBP1(U) to that detected in the vector-co-transfected cells (compare 7 with lane 6). In contrast, ATF4 levels were not affected by co-expression of pXBP1(U) (Fig. 3A, lower panel, compare lane 2 with lane 1) but were markedly increased by treatment with MG132 (compare lane 6 with lane 1). These results suggest that pXBP1(U) targets ATF6 but not ATF4 in proteasome-mediated degradation.
 View Details | Fig. 3. Effect of co-expression of full-length or various deletion mutants of pXBP1(U) on expression levels of ATF6 and ATF4. (A, B) HeLa cells were transfected with pcDNA-ATF6α(N) (A, upper panel) or pcDNA-ATF4 (A, lower panel) together with a 9-fold excess of pcDNA alone (vector), or full-length or various deletion mutants of pcDNA-XBP1(U) as indicated at top. Transfected cells were treated (+) or untreated (–) with 10 μM MG132 for 2 h prior to harvest. Lysates were prepared and analyzed by immunoblotting using anti-ATF6α (A, upper panel), anti-ATF4 (A, lower panel) or anti-XBP1 antibody (B). Asterisks denote non-specific bindings. [A and B, bottom panels] Intensity of each band was quantified and band intensity of ATF6α or XBP1 in cells treated without MG132 was calculated relative to that with MG132 (taken as 100%) for each transfection. Data are presented as the means of two independent experiments with error bars. |
We recently showed that rapid degradation of pXBP1(U) by the proteasome is mediated by the DEG present in the C-terminal tail region of pXBP1(U), and that deletion mutants of pXBP1(U) lacking the DEG are therefore stable (Yoshida et al., 2006). This notion is represented in Fig. 3B. We also found that co-expression of pATF6α(N) or ATF4 did not affect these properties. Thus, levels of full-length pXBP1(U) were increased by MG132 treatment because proteasome-mediated degradation was blocked (compare lane 7 with lane 2), whereas those of the three deletion mutants of pXBP1(U) lacking the DEG were not (compare lanes 8–10 with lanes 3–5). Importantly, none of the three deletion mutants of pXBP1(U) lacking the DEG decreased the level of co-expressed pATF6α(N), in contrast to the case of full-length pXBP1(U) (Fig. 3A, upper panel, compare lanes 3–5 with lane 1). Rather, the level of pATF6α(N) was increased in cells co-expressing the deletion mutants of pXBP1(U) lacking the DEG, especially pXBP1(U)[1–133], as compared with that in vector-transfected cells (Fig. 3A, upper panel, compare lane 4 with lane 1, see below for possible explanation). These results indicate the importance of the DEG in not only the degradation of pXBP1(U) but also the degradation of pATF6α(N) induced by co-expression of pXBP1(U).
To confirm whether co-expression of pXBP1(U) affected the stability of pATF6α(N), cycloheximide chase experiments were performed. Results showed that degradation of pATF6α(N) (Fig. 4A, lanes 1–3) was indeed accelerated by co-expression of full-length pXBP1(U) (lanes 4–6) but not by a deletion mutant of pXBP1(U) lacking the DEG and NES (lanes 7–9), namely pXBP1(U)[1–185]. Consistent with the results shown in Fig. 3A, the level of pATF6α(N) in cells co-expressing pXBP1(U)[1–185] was higher than that in cells expressing vector alone (compare lane 7 with lane 1). Because pXBP1(U)[1–133] and pXBP1(U)[1–185] are exclusively localized in the nucleus (Yoshida et al., 2006), they might have stabilized pATF6α(N) by antagonizing endogenous pXBP1(U)-mediated degradation of pATF6α(N), leading to increased stability of pATF6α(N), as observed in Fig. 4A (compare lanes 7–9 with lanes 1–3), as well as enhanced accumulation of pATF6α(N), as observed in Fig. 3A (compare lane 4 with lane 1) and Fig. 4A (compare lane 7 with lane 1).
 View Details | Fig. 4. Effect of co-expression of full-length or a deletion mutant of pXBP1(U) on stability and localization of ATF6. (A) HeLa cells were transfected with pcDNA-ATF6α(N) together with a 9-fold excess of pcDNA vector alone, pcDNA-XBP1(U) or pcDNA-XBP1(U)[1–185]. Transfected cells were treated with 40 μM cycloheximide for the indicated periods. Lysates were prepared and analyzed by immunoblotting using anti-ATF6α antibody. Asterisks denote non-specific bands. Lower panel shows longer exposure of the film for better comparison of stability. [bottom panels] Intensity of each band was quantified and normalized with the value at 0 h. Data are presented as the means of two independent experiments with error bars. (B) HeLa cells were transfected as in (A). Transfected cells were fixed and stained with anti-ATF6α antibody. Cells were also counterstained with DAPI. |
We then examined whether the co-expression of pXBP1(U) affected the localization of pATF6α(N). pATF6α(N) was localized exclusively in the nucleus (Fig. 4B, panel a), but was mostly present in the cytoplasm in cells co-expressing full-length pXBP1(U) (panel d). pATF6α(N) was again localized in the nucleus in cells co-expressing a deletion mutant of pXBP1(U) lacking both the DEG and NES (panel g). These results suggest that pXBP1(U) causes relocation of pATF6α(N) from the nucleus to the cytoplasm via NES activity, and induces proteasome-mediated degradation of pATF6α(N) via the action of the DEG, both being present in the C-terminus of pXBP1(U).
We finally examined whether the stability of endogenous pATF6α(N) was affected by the presence or absence of endogenous pXBP1(U), using wild-type (XBP1+/+) and XBP1-deficient (XBP1–/–) MEFs. The cells were first treated with dithiothreitol, a reducing reagent known to strongly induce ATF6 proteolysis (Nadanaka et al., 2007), for 2 h to produce pATF6α(N). The dithiothreitol was then washed out to block subsequent cleavage of ATF6α, allowing us to check the subsequent stability of pATF6α(N). It was previously shown that transcriptional induction of only a subset of ER chaperones is compromised in XBP1–/– MEFs and thus the levels of major ER chaperones such as BiP and GRP94 before and after ER stress are not affected by the absence of XBP1 (Lee et al., 2003), indicating that the ER of XBP1 +/+ and –/– MEFs is stressed similarly by dithiothreitol treatment. Indeed, as shown in Fig. 5A, pATF6α(P), a precursor form of ATF6α, was cleaved similarly in XBP1 +/+ and –/– MEFs, and comparable amounts of pATF6α(N) were produced after 2-h treatment with dithiothreitol (lanes 2 and 7 and lanes 12 and 17). Importantly, pATF6α(N) was decreased in XBP1+/+ MEFs (lanes 2–5 and lanes 12–15) but not in XBP1–/– MEFs (lanes 7–10 and lanes 17–20) (see quantification data below). In contrast, no significant difference was seen in the levels of ATF4 between XBP1+/+ and XBP1–/– MEFs, (compare lanes 2–5 with 7–10 and lanes 12–15 with 17–20), although levels were not decreased but rather increased after dithiothreitol wash out. A likely explanation is that it takes more time to stop the production of ATF4 than of pATF6α(N), because the production of pATF6α(N) is a direct consequence of ER stress-induced activation of ATF6α, whereas that of ATF4 is not a direct consequence of ER stress-induced activation of upstream PERK; rather, ATF4 is translationally induced via PERK-mediated phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (Harding et al., 2000), and the dissociation of ATF4 mRNA from the polyribosome would therefore take some time. pXBP1(S) and pXBP1(U) were hardly detected under these conditions, probably because our anti-human XBP1 antibody recognizes mouse XBP1 only poorly. To facilitate detection, MEFs were treated with MG132 before and after dithiothreitol treatment and after wash out. As shown in Fig. 5B, pXBP1(U) but not pXBP1(S) was constitutively detected in XBP1 +/+ (lane 1) but not in XBP1–/– (lane 6) MEFs, as expected. Both pXBP1(U) and pXBP1(S) were detected after treatment with dithiothreitol (lane 2), and levels were increased after wash out owing to the blockade of degradation by MG132 (lanes 3–5). We concluded that endogenous pXBP1(U) is involved in the degradation of endogenous pATF6α(N) but not ATF4.
 View Details | Fig. 5. Effect of the presence or absence of XBP1 on degradation of ATF6 and ATF4. (A) XBP1+/+ and XBP1–/– MEFs were treated with 1 mM dithiothreitol (DTT) for 2 h. DTT was then washed out and the cells were incubated for the indicated periods in the absence of DTT. Cell lysates were prepared and analyzed by immunoblotting using anti-ATF6α and anti-ATF4 antibodies. Results of two independent experiments are shown. Asterisks denote non-specific bands. [bottom panels] Intensity of each band was quantified, normalized with the value at 0 min in XBP1 +/+ MEFs, and plotted against incubation periods as the means with error bars. (B) XBP1+/+ and XBP1–/– MEFs were treated as in (A) except that 10 μM MG132 was included in the culture from 1 h prior to the addition of DTT until the end of incubation in the absence of DTT. Cell lysates were prepared and analyzed by immunoblotting using anti-XBP1 antibody which detects both pXBP1(S) and pXBP1(U). |
Three UPR regulators are activated in mammalian cells to counter ER stress, namely IRE1, PERK and ATF6. XBP1 mRNA, a substrate of IRE1-mediated unconventional splicing, is constitutively expressed albeit at a low level, and translated to produce pXBP1(U) (Calfon et al., 2002, Yoshida et al., 2001). This is in marked contrast to its yeast counterpart, HAC1 mRNA, which is constitutively expressed at a relatively high level but not translated (Chapman and Walter, 1997; Kawahara et al., 1997). We recently showed that the expression level of pXBP1(U) is elevated in the recovery phase of ER stress, leading to the enhanced degradation of pXBP1(S), a transcriptionally active XBP1 produced from spliced XBP1 mRNA, and the shut-off of remnant transcription of XBP1-target genes, such as EDEM, a component of the ERAD machinery (Yoshida et al., 2006). Here, we found that pXBP1(U) specifically binds to pATF6α(N) and renders it more susceptible to the proteasome, suggesting that pXBP1(U) negatively regulates the activity of pATF6α(N) to stop residual transcription of ER chaperone and ERAD component genes during the recovery phase. Common negative regulation mediated by pXBP1(U) would be beneficial to the cell as pATF6α(N) and pXBP1(S) are known to cooperate; we have previously shown that pATF6α(N)-pXBP1(S) heterodimer mediates the induction of ERAD component genes in response to ER stress (Yamamoto et al., 2007). By allowing pXBP1(U) to down regulate both pATF6α(N) and pXBP1(S) simultaneously, the cell can shut off transcription of ERAD component genes rapidly and efficiently when ER stress is halted, so that newly synthesized proteins in the absence of ER stress would be maximally utilized for the cell with less chance of being degraded by mistake.
As we reported for the degradation of pXBP1(S) (Yoshida et al., 2006), pXBP1(U)-induced degradation of pATF6α(N) is mediated by the DEG located at the pXBP1(U)-specific C-terminal end. It remains to be determined how the DEG mediates degradation of pXBP1(S) and pATF6α(N). The DEG may be a binding site of a specific E3 ubiquitin ligase. Interestingly, however, pXBP1(U) neither associated with nor enhanced the degradation of ATF4, a key transcription factor of the PERK pathway which is capable of activating the transcription of genes involved in amino acid metabolism and anti-oxidative stress (Harding et al., 2003). Our finding is consistent with the results of a previous study which examined the interaction between 49×49 pairs of bZIP transcription factors in vitro, and found that XBP1 dimerized with itself and ATF6 but not ATF4 (Newman and Keating, 2003). Thus, we concluded that pXBP1(U) is a negative feedback regulator specific to the ATF6 and IRE1 branches of the mammalian UPR. This also indicates the presence of cross-talk between the IRE1 and ATF6 pathways. We previously identified another example of cross-talk between these pathways, namely the activation of transcription of the XBP1 gene by pATF6(N) through the ER stress response element present in the XBP1 promoter (Yoshida et al., 2000). These findings suggest that the ATF6 and IRE1 pathways are interactive, while the PERK pathway may be relatively independent.
There appear to be several explanations for why pXBP1(U) does not affect ATF4. First, ATF4 is not a UPR-specific transcription factor, but is rather involved in various stress responses such as amino acid starvation, viral infection and heme deficiency, which are collectively called the integrated stress response (Harding et al., 2003). If pXBP1(U) negatively regulated ATF4, expression of ATF4 would be mitigated during the integrated stress response, since a low level of pXBP1(U) is constitutively produced in cells unexposed to ER stress (Yoshida et al., 2001; Yoshida et al., 2006). Second, ATF4 has its own feedback mechanism: ATF4 induces expression of GADD34 which, in conjunction with protein phosphatase 2C, dephosphorylates the α subunit of eukaryotic initiation factor 2, which is responsible for ER stress-induced translational attenuation, leading to blockade of the translational induction of ATF4 (Novoa et al., 2001). Third, the prolonged expression of genes for amino acid metabolism and anti-oxidative stress, the targets of ATF4, might be less toxic than that of genes for ER chaperones and ERAD components, and thus a need for rapid turn-off of the remaining transcription may not be required.
Fig. 1 shows the functional domains of pXBP1(U) and pXBP1(S) identified in the present and previous studies. Both molecules share the N-terminal region, b (functions as nuclear localization signal and DNA-binding domain) and ZIP domains. pXBP1(U) contains the NES and DEG, whereas pXBP1(S) has an AD. Thus, XBP1 consists of elaborately designed modules, and its function is radically switched by unconventional mRNA splicing initiated by IRE1. Since this mRNA splicing is thought to take place in the cytoplasm, it is called “cytoplasmic splicing” (Back et al., 2006; Ruegsegger et al., 2001; Yoshida et al., 2006). Why have mammalian cells adopted cytoplasmic splicing for the UPR, instead of the conventional nuclear splicing? We speculate that cytoplasmic splicing has an advantage over nuclear splicing, in that IRE1 senses ER stress by its luminal domain, and directly cleaves XBP1 pre-mRNA by its ribonuclease domain, which is present in the cytoplasmic region. In this way, cytoplasmic splicing can switch the function of XBP1 from pXBP1(U), a negative regulator, to pXBP1(S), a positive regulator, in response to ER stress much faster than could be achieved by nuclear splicing. At present, mammalian XBP1 and yeast HAC1 mRNAs are the only known substrates of cytoplasmic splicing. Identification and analysis of further as yet unknown substrates will advance our understanding of the biological significance of cytoplasmic splicing.
We are grateful to Dr. David Ron (New York University) for providing ATF4 expression plasmid and Dr. Laurie Glimcher (Harvard Medical School) for providing XBP1+/+ and XBP1–/– MEFs. We thank Ms. Kaoru Miyagawa for technical and secretarial assistance. This work was supported, in part, by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (18050013, 19370086 and 20052014 to H. Y. and 15GS0310 and 19058009 to K. M.).
|