| To whom correspondence should be addressed: Koji Ikegami, Department of Molecular Anatomy, Molecular Imaging Advanced Research Center, Hamamatsu University School of Medicine, Hamamatsu, Shizuoka 431-3192, Japan. Tel: +81–53–435–2085, Fax: +81–53–435–2292 E-mail: kikegami@hama-med.ac.jp Abbreviations: mAb, monoclonal antibody; MAP, microtubule-associated protein; MT, microtubule; PTM, post-translational modification; TTL, tubulin tyrosine ligase. |
The microtubule (MT) is one of three major cytoskeletons highly conserved in eukaryotes. As the name indicates, the MT has a tubular structure with a diameter of 25 nm (Fig. 1). The tubule wall is composed of thirteen protofilaments, which is further constructed by heterodimers of two small globular proteins, α- and β-tubulins (Mohri, 1968) (Fig. 1). MTs play specific roles in a variety of cellular events, which range from directional molecular transport, ciliary or flagellar motility, and chromosome segregation, to cytokinesis. To play these specialized roles, the MT constructs higher-order architecture in related subcellular compartments, such as bundles (in neuronal processes), axonemes (in flagella and cilia), and centrioles (in basal bodies or spindle bodies) (Fig. 1).
 View Details | Fig. 1. Microtubule in cell. Microtubule-enriched subcellular compartments or structures are depicted. Microtubule is shown in red. Microtubule-constructed higher architectures, such as bundle, axoneme, and centriole, are highlighted in boxes. Individual microtubule with diameter of 25 nm is composed of 13 protofilaments, which is further constructed from multiple heterodimers of α- and β-tubulins. PTMs currently known are listed below. |
How does the simple tubular MT form the highly organized architecture? The mechanism relies on interactions between MT and a variety of its binding partners. The molecules contain various proteins with a wide range of mass, structure, and function. Molecular motor proteins, kinesins and dyneins, use MT as a molecular railway and walk on it. MT-associated proteins (MAPs) starch up the MTs, and thus stabilize them. Plus-end tracking proteins (+TIPs) accumulate on the distal tip of the MT. Some other molecular species can sever and thus destabilize MT. Those MT-interacting proteins associate with MTs in designated subcellular positions to play their roles properly.
What leads the MT-interacting proteins to their work place and how are they regulated, as different kinesin motors deliver their cargos to each specific destination in a neuronal cell (Setou et al., 2000; Setou et al., 2002; Setou et al., 2004)? Two mechanisms can be assumed. One is a well-known regulatory system, the post-translational modifications (PTMs) of MT-interacting proteins. The most popular example is the phosphorylation of MAPs. Evidence has accumulated that the phosphorylation of MAPs affects their function. The phosphorylation of tau inhibits the function of tau to promote MT assembly (Nishida et al., 1982; Yamamoto et al., 1983; Lindwall and Cole, 1984; Hoshi et al., 1987; Wada et al., 1998). The phosphorylation of MAP2 counteracts MAP2-mediated microtubule polymerization (Jameson et al., 1980; Nishida et al., 1981; Yamamoto et al., 1983; Nishida et al., 1987; Hoshi et al., 1988). Hence, phosphorylation of MAPs negatively regulates MTs dynamics and stability (Drewes et al., 1997). CRMP2 phosphorylation inactivates the activity of CRMP2 to promote microtubule assembly and controls neuronal polarity (Yoshimura et al., 2005). Moreover, MAPs phosphorylation indirectly modulates kinesin motor trafficking through affecting MAPs-MT binding affinity (Sato-Harada et al., 1996).
The multi-microtubule hypothesis can support another mechanism. The heterogeneities of tubulin molecules underlie the model (Kobayashi and Mohri, 1977). The heterogeneities are made by two independent mechanisms: genome-encoded multi-tubulin genes and PTMs. Mammalian genome encodes 6 to 8 different tubulin molecules for each α- and β-tubulins. Those different tubulins show different tissue-specific expression patterns. PTMs can more drastically contribute to the heterogeneities.
Tubulin is subjected to the detyrosination/tyrosination cycle, the removal of penultimate glutamate, polyglutamylation, polyglycylation, acetylation, phosphorylation, and palmitoylation (Fig. 1). These modifications, except for acetylation, occur in the C-terminal region, which surfaces on MT lattice where many MT-binding proteins interact. In this review, we focus on unique PTMs (detyrosination/tyrosination, polyglutamylation, and polyglycylation), and review the currently available evidence for the roles of the PTMs in mammals with emphasis on recent progress. We also present some evidence obtained from genetically modified non-mammalian organisms. Readers are referred to some review articles for other general modifications (acetylation, phosphorylation, and palmitoylation) (Rosenbaum, 2000; Westermann and Weber, 2003; Verhey and Gaertig, 2007; Hammond et al., 2008).
The cycle of detyrosination and tyrosination is a PTM of tubulin that has been investigated for the longest time. This PTM occurs in most α-tubulins. In this cycle, a tyrosine that is encoded as the C-terminal amino acid by genome is removed (Argaraña et al., 1978), and a free tyrosine is re-added to the C-terminal of detyrosinated α-tubulin (Barra et al., 1974). Long-living, i.e. stable MTs are enriched by detyrosinated tubulins while highly dynamic MTs contain abundantly tyrosinated tubulins (Wehland and Weber, 1987). A recent work demonstrates that tubulin detyrosination inhibits MT disassembly through suppressing interactions of MT-depolymerizing motor, MCAK or KIF2 with MTs (Peris et al., 2009), although detyrosination has been thought not to modify MT stability for a long time (Webster et al., 1990). Detyrosinated tubulin is further subjected to next deglutamylation, i.e. removal of penultimate glutamate, producing Δ2-tubulin (Paturle-Lafanechère et al., 1991) (Fig. 1). In the current model, Δ2-tubulin is thought to be excluded from the detyrosination/tyrosination cycle (Westermann and Weber, 2003; Rüdiger et al., 1994).
An enzyme that accounts for the detyrosination/tyrosination cycle is the oldest enzyme identified as a tubulin-modifying enzyme. The enzyme for re-addition of tyrosine to detyrosinated α-tubulin, termed tubulin tyrosine ligase (TTL) (Raybin and Flavin, 1977) (Fig. 2), is purified from porcine brain (Murofushi, 1980), and the polypeptide is completely identified (Ersfeld et al., 1993). In contrast, the enzyme removing the terminal tyrosine is not fully identified, while recent researches propose a cytosolic carboxy peptidase, CCP1/Nna1 as a strong candidate (Kalinina et al., 2007; Rodriguez de la Vega et al., 2007) (Fig. 2). Enzymes performing the removal of penultimate glutamate from detyrosinated tubulin remain to be identified. Enzymes re-adding glutamate to Δ2-tubulin might be present, though currently nobody suggests this idea.
 View Details | Fig. 2. Enzyme for tubulin PTMs. Identified enzymes for modifications are shown in red boldface. Carboxypeptidase for generating detyrosinated tubulin is still only a candidate (yellow). Unidentified enzymes are shown in light gray. |
Recent progress for understanding the physiological roles of this modification cycle is achieved by a generation of a TTL-null mouse line. The TTL-null animal provides evidence for a vital role of tyrosinated tubulin. The mouse shows malformation of layer in cerebral cortex, disruption of cortico-thalamic loop, and perinatal lethality along with prominent loss of tyrosinated tubulins (Erck et al., 2005). Neurons cultured from the animal display abnormal neurite growth along with forming multiple axons (Erck et al., 2005). In the neurons, the cytoplasmic linker protein (CLIP)-170, a +TIP containing glycine-rich cytoskeleton-associated protein (CAP-Gly) domains, is dispersed from the growth cone (Erck et al., 2005). Fibroblasts cultured from the TTL-null animal display mislocalization of CLIP-170 and abnormal orientations of spindle bodies (Peris et al., 2006). Indeed, CLIP-170 preferentially binds in vitro to tyrosinated α-tubulin (Peris et al., 2006) and to the –EEY/F motif seen in the α-tubulin C-terminal (Honnappa et al., 2006; Mishima et al., 2007). Similar results are observed in a most primitive eukaryote, the budding yeast. Genetical deletion of phenylalanine from the C-terminal of α-tubulin results in mislocalization of Bik1p, a yeast homolog of CLIP-170 (Badin-Larçon et al., 2004).
Another line of evidence indicates that the modification affects the interaction between molecular motors and MTs. Kinesin-1 shows more strong binding affinity to MT enriched by detyrosinated tubulin in vitro (Liao and Gundersen, 1998). In vivo, kinesin-1 preferentially binds to detyrosinated tubulin-rich MTs (Dunn et al., 2008). Kinesin-1 selectively accumulates in a future axon in early stage of neuronal growth (Jacobson et al., 2006). The axonal shaft is more enriched by detyrosinated tubulins than dendrites and cell body during early neuronal differentiation (Arregui et al., 1991). In light of the knowledge that has accumulated, our group has recently provided clear evidence that tubulin detyrosination is a spatial cue for neuronal polarity determination through preferential interaction between KIF5-loop8 and detyrosinated tubulin-rich MTs (Konishi and Setou, 2009). The reason why neurons cultured from the TTL-null animal form multiple axons (Erck et al., 2005) can be explained by this model, whereby kinesin-1 might fail to find the future axon since MTs are uniformly detyrosinated. Such a model allows us to speculate that the neuronal degeneration observed in Purkinje cell degeneration (pcd) mouse (Mullen et al., 1976; Landis and Mullen, 1978), a spontaneous mutant of CCP1/Nna1 (Fernandez-Gonzalez et al., 2002), might result from inefficient intracellular transports caused by lowered binding affinity between kinesin-1 and MTs that lack detyrosinated tubulins.
Tubulin polyglutamylation was first reported in pig brain MT (Eddé et al., 1990). This modification is highly intriguing from two points. First, the form of modification is unique. A long chain of polypeptides constructed from multiple glutamates are covalently linked on a γ-carboxy group of glutamate residue in tubulin C-terminal region (Fig. 3). Second, the long polyglutamate chain increases remarkably negative charges in the tubulin C-terminal (Fig. 3), which readily suggests the important roles of the charges in the interaction between MT and its binding partners. Studies using a specific monoclonal antibody (mAb) GT335 against glutamylated tubulin reveal that polyglutamylated tubulins are enriched in neuronal cells (Wolff et al., 1992), and that axonemal and centriolar MTs are also highly modified (Fouquet et al., 1994; Bobinnec et al., 1998b).
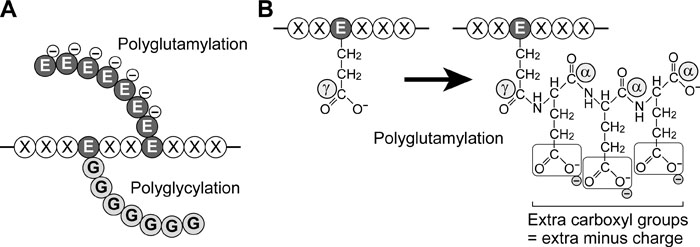 View Details | Fig. 3. Polyglutamylation and polyglycylation. (A) Schematic view of polyglutamylation and polyglycylation. Poly-glutamate (EEE···) or -glycine (GGG···) chain is attached to a glutamate residue (E) in a protein backbone. Note that the polyglutamate chain has negative charges. (B) More detailed diagram of polyglutamylation. The first glutamate is bound to a γ-carboxyl group of a glutamate residue. Subsequent addition of glutamates is achieved by bonds between α-carboxyl group and amino group. Note that the polyglutamate chain provides extra carboxyl groups that result in increases in total negative charges. |
A recent significant breakthrough in the area of polyglutamylation research is the success in identifying modification-performing enzymes, polyglutamylases (or glutamate ligases) (Fig. 2). Interestingly, the enzymes are members of a protein family homologous to TTL. The first identified TTLL1 is a subunit of the polyglutamylase complex purified from mouse brain (Janke et al., 2005). Further studies demonstrate that TTLL4, 5, 6, 7, 9, 11, and 13 have tubulin polyglutamylase activity (Ikegami et al., 2006; van Dijk et al., 2007; Wloga et al., 2008; Mukai et al., 2009). Currently, the glutamate-removing enzyme, deglutamylase or glutamatase remains to be identified. CCP 2 to 6, which are members of cytosolic carboxypeptidases with similarity to CCP1/Nna1 (Kalinina et al., 2007; Rodriguez de la Vega et al., 2007), might harbor the enzymes.
The identification of enzymes enables us to better understand the physiological roles of tubulin polyglutamylation in detail. A genetically manipulated mouse line, which lacks PGs1 (Campbell et al., 2002), a component of the TTLL1-containing polyglutamylase complex (Regnard et al., 2003; Janke et al., 2005), provides strong evidence. The PGs1-deficient mouse shows impaired synaptic transmission and a decrease in the amount of synaptic vesicles in synaptic terminals associated with a gross loss of polyglutamylation on α-tubulin in brain homogenate (Ikegami et al., 2007). Trafficking of KIF1A (kinesin-3 family), but neither KIF3A (kinesin-2 family) nor KIF5A (kinesin-1 family), is impaired in the neurons cultured from the mouse (Ikegami et al., 2007). In vitro, binding of KIF1A to MT is most affected by the loss of polyglutamylated α-tubulin (Ikegami et al., 2007). This KIF1-selective impairment can be explained by the finding that KIF1 has a longer K-loop than KIF3 or KIF5 (Okada and Hirokawa, 1999).
Evidence has accumulated for the spatial and temporal distribution of polyglutamylated tubulins. In neuronal cells, polyglutamylated β-tubulin concentrates in the somato-dendritic region, whereas polyglutamylated α-tubulin accumulates in the neuronal processes (Ikegami et al., 2006; Ikegami et al., 2007) (Fig. 4). In mouse brain, polyglutamylated β-tubulin emerges after birth, whereas modified α-tubulin is already prominent at birth (Audebert et al., 1994). In culture, polyglutamylated β-tubulin emerges and increases in parallel with neuronal growth, while modified α-tubulin is detected immediately after starting the culture (Audebert et al., 1994). The late increase in level of polyglutamylated β-tubulin may be involved in postnatal growth and maturation of neuronal processes, especially dendrites. Indeed, knockdown of TTLL7 in PC12 cells demonstrates that TTLL7-mediated β-tubulin polyglutamylation is required for neurite growth (Ikegami et al., 2006). What involves polyglutamylated β-tubulin in neurite growth currently remains to be elucidated. However, MAP1A is a plausible candidate. This hypothesis can be supported by three independent lines of evidence: i) strong dependency of MAP1A-MT interaction on level of tubulin polyglutamylation (Bonnet et al., 2001; Ikegami et al., 2007); ii) postnatal increase in level of MAP1A in developing mouse brain (Schoenfeld et al., 1989); iii) the important role of MAP1A in activity-dependent dendritic growth and branching (Szebenyi et al., 2005).
 View Details | Fig. 4. Distribution of polyglutamylated and polyglycylated tubulins. Note that the subunits of tubulin that are well polyglutamylated are different between somato-dendrites and axons. Currently, no evidence is available that polyglycylated tubulin is detected in centrioles. |
The roles of tubulin polyglutamylation in flagella or cilia are still subject to controversy. The PGs1-deficient mouse shows malformation of sperm flagella, but not cilia in trachea, nasal duct, or oviduct (Campbell et al., 2002), suggesting that tubulin polyglutamylation is required for axoneme construction and that the modification has distinct roles in flagella and cilia. Studies where modification-specific antibodies are used provide supportive evidence. Ciliary motility of human respiratory epithelium is significantly inhibited by injection of mAb GT335 (Million et al., 1999). A similar result is observed in a work where sea urchin sperm is used. The flagellar motility was affected by injection of another mAb B3 into the sperm (Gagnon et al., 1996). Although the numerous studies employing antibodies indicate the importance of tubulin polyglutamylation in ciliary or flagellar motility, Tetrahymena, a ciliated protozoan swims normally even if it lacks both TTLL1 and TTLL9 (Wloga et al., 2008). The role of tubulin polyglutamylation may differ in organism species. To resolve these questions, we anticipate the development of mammalian models, the TTLLs of which are genetically modified.
A recent work investigating zebrafish provides evidence for broad function of tubulin polyglutamylation in the cilia of the whole body. A knockdown of Fleer, a newly identified regulator of tubulin polyglutamylation, or TTLL6 results in structural and functional abnormalities throughout the whole body (Pathak et al., 2007). However, the function of the modification in centrioles is blurrier. The only evidence available is that mAb GT335 destabilizes the centrioles when injected into living cells (Bobinnec et al., 1998a). To achieve a more direct and accurate understanding requires more robust investigations, e.g. those examining polyglutamylation-performing TTLL knockout animals.
Polyglycylated tubulin is abundantly detected in axonemal MTs (Redeker et al., 1994) (Fig. 4). In polyglycylation, a poly-glycine chain is bound to a glutamate residue in tubulin C-terminal region. In contrast to polyglutamylation, polyglycylation does not alter the net charge of the tubulin C-terminal (Fig. 3). This simple addition of a mere polypeptide that has no outstanding characteristic had made it difficult for researchers to anticipate the function of the modification. However, the roles of tubulin polyglycylation are now beginning to be unveiled, as TTLLs 3, 8, and 10 are identified as polyglycylation-performing enzymes, the polyglycylases (or glycine ligases) (Ikegami et al., 2008; Ikegami and Setou, 2009; Rogowski et al., 2009; Wloga et al., 2009b).
While understanding the physiological roles of the tubulin polyglycylation in mammals has been slow to emerge, elegant works using Tetrahymena have provided interesting evidence for the roles of tubulin polyglycylation. Mutations in polyglycylation sites of β-tubulin, but not of α-tubulin, have been reported to impair cellular motility and cytokinesis (Xia et al., 2000). Ultrastructurally, the axonemes of the mutant lose the central-pair MTs and B-subfibers (Thazhath et al., 2002), suggesting that tubulin polyglycylation has a pivotal role in constructing ciliary axonemes. More recently, it has been revealed that tubulin polyglycylation by TTLL3 is essential for proper cilia assembly in Tetrahymena and zebrafish (Wloga et al., 2009b). Interestingly, TTLL3-deficient Tetrahymena shows a counter-increase in polyglutamylation along with a decrease in tubulin polyglycylation (Wloga et al., 2009b), which is consistent with the concept of cross-talk of PTMs in α- and β-tubulin C-termini (Redeker et al., 2005). The increase in polyglutamylation could explain the cause of abnormal cilia formation, as over-polyglutamylation, manipulated by the overexpression of the polyglutamylase TTLL6, destabilizes axonemal microtubules (Wloga et al., 2009a). These findings can provide a cue for understanding the molecular machinery whereby tubulin polyglycylation performs its function. The MT-severing enzyme katanin might be one of the machineries since the phenotypes of the mutant MT are copied in katanin-deficient organism (Sharma et al., 2007), though it remains unclear whether or not katanin recognizes polyglycylation or polyglutamylation. Given these intriguing clues, the development of genetically manipulated mammalian models are now eagerly awaited.
Our understanding of the unique tubulin PTMs has proceeded dramatically during the past few years. The recent advances are summarized in Table I. The progress achieved is due in large part to the identification of modification-performing enzymes and the development of genetically manipulated organisms. Research on the roles of tyrosination/detyrosination cycle are becoming ever more sophisticated. The identification of polyglutamylases and polyglycylases has enabled research studies in the field of polyglutamylation and polyglycylation to enter the next stage. Function in mammals can be inferred from studies employing more primitive organisms, like small ciliates, which are easily manipulated genetically. Findings obtained from gene-knockdown in zebrafish have provided valuable information to anticipate the functions of PTMs in mammals. Nonetheless, more direct evidence is expected to be obtained from genetically manipulated mice. Those animals will enable us to better understand the physiological function of the modifications more directly. The additional and multiplicative effects of other PTMs will be a major theme in future works. In certain cases, the coordinated or competitive regulation of PTMs will be a problem. Spatio-temporal dynamic regulation of multiple PTMs will be the next frontier. It is amply clear that we are standing on the threshold of a vast field of studies on tubulin PTMs.
This review was written as part of the Young Scientist Award for Best Presentation of the Japan Society for Cell Biology awarded to Koji Ikegami at the 61st JSCB Annual Meeting. The authors would like to express their heartfelt thanks to Prof. Kozo Kaibuchi, the chair of the 61st JSCB Annual Meeting, and the organizing committee of the 61st JSCB Annual Meeting for giving us this opportunity to write this review. We are also grateful to the present and former members of the Setou laboratory of the Hamamatsu University School of Medicine and the Mitsubishi Kagaku Institute of Life Sciences for their technical assistance, valuable discussions, and encouragement.