| To whom correspondence should be addressed: Hiderou Yoshida, Graduate School of Life Science, University of Hyogo, Harima Science Garden City, Hyogo 678-1297, Japan. Fax: +81–791–58–0212, Tel: +81–791–58–0219 E-mail: hide@sci.u-hyogo.ac.jp http://www.sci.u-hyogo.ac.jp/life/biochem2/index-j.html |
The Golgi apparatus is an organelle in which secretory proteins undergo various kinds of processing, such as modification of sugar moieties and cleavage of peptide bonds, and are sorted to their final destinations. Interestingly, the Golgi apparatus is well developed in cells secreting a large amount of proteins. For instance, the secretory mucous cells of Brunner’s gland have an extensive Golgi apparatus (Berger and Roth, 1997). Moreover, the amount of the Golgi apparatus is dynamically changed in prolactin cells of the pituitary glands and in acinar cells of the mammary glands of female mice in response to the suckling stimulus, which enhances secretion of prolactin and milk proteins (Clermont et al., 1993; Rambourg et al., 1993). These observations suggest that the functional capacity of the Golgi apparatus is tightly regulated in accordance with cellular needs, though the underlying mechanism has not been clarified (Yoshida, 2009).
In the case of the endoplasmic reticulum (ER), the mechanism called the ER stress response (or the unfolded protein response) adjusts the ER capacity to the cellular demands (Kohno, 2010; Mori, 2009; Ron and Walter, 2007; Scheuner and Kaufman, 2008; Yoshida, 2007a). If cells produce a large amount of secretory proteins in the ER and the capacity of the ER is overwhelmed, nascent proteins cannot be properly folded by ER chaperones, and unfolded proteins are accumulated in the ER (ER stress). Sensor molecules located on the ER membrane, such as pATF6(P), IRE1 and PERK, detect ER stress and induce the expression of active transcription factors such as pATF6(N), XBP1 and ATF4, which increase the transcription of ER-related genes, including ER chaperones and ER-associated protein degradation machinery, to upregulate the capacity of the ER. Actually, overexpression of XBP1 or ATF6 induces ER expansion (Bommiasamy et al., 2009; Sriburi et al., 2004), and the ER of secretory cells, such as plasma cells, pancreatic and salivary gland acinar cells, is poorly developed in XBP1-knock out mice (Lee et al., 2005; Reimold et al., 2001), suggesting that these transcription factors are essential for upregulation of the ER capacity.
If the capacity of ER function were increased by the ER stress response, large amounts of secretory proteins would be transported to and inundate the Golgi apparatus, and thus most secretory proteins could not be processed by Golgi-resident proteins, including Golgi-specific glycosylation enzymes, since the amount of secretory proteins would surpass the capacity of these enzymes. Cells have to cope with such a stressful situation (Golgi stress) by upregulating the capacity of the Golgi apparatus. Since the genes involved in the function of the Golgi apparatus are located in the nucleus, the signal of Golgi stress detected by yet unknown sensors has to be transmitted from the Golgi to the nucleus, where the transcription of Golgi-related genes is increased to augment the Golgi capacity. Identification of such a regulatory circuit controlling the capacity of the Golgi apparatus in accordance with cellular demands, which we have named the Golgi stress response, would contribute to solving a fundamental problem in cell biology, that is, how the capacity of each organelle is regulated in the cell. In this study, we explored the possibility of the existence of the Golgi stress response in mammalian cells, and identified a cis-acting element regulating transcriptional induction by the Golgi stress response.
Culturing and transient transfection of HeLa cells were carried out as described previously (Yoshida et al., 2003). After transfection, cells were treated with or without 0.3 μM monensin (Calbiochem) or 1 μM nigericin (Calbiochem) for 16 h, washed three times with PBS, incubated in fresh medium for 6 h and harvested for immunoblotting or luciferase assays. For Northern or Western blot analysis, cells were treated with 10 μM monensin, whereas they were treated with 0.3 μM monensin for luciferase assays, since we later found that treatment with 0.3 μM monensin is sufficient to induce transcriptional induction. For filipin staining, cells were fixed with formaldehyde and incubated with 0.05 mg/ml filipin (Sigma) for 2 h. Fluorescent microscopy was carried out with a TE2000-U (Nikon) and ORCA-ER (Hamamatsu Photonics).
Total RNA was extracted from HeLa cells treated with or without 10 μM monensin for 8 h by the acid guanidium/phenol/chloroform method using Sepasol (Nacalai Tesque), and purified using RNeasy Mini column (Qiagen). The purified RNA was checked for quality with an Agilent 2100 Bioanalyzer (Agilent Technologies), converted to cDNA, and labeled with cyanine 3-CTP and cyanine 5-CTP. Human 1A Oligo Microarray version 2 (Agilent Technologies) on which 20,173 human genes were spotted was probed with these labeled probes. Genes showing fluorescent intensity lower than average background intensity for all array spots were eliminated from further analysis. Finally, we analyzed 18,062 human genes that showed sufficient signal intensity in all four independent array experiments.
To construct reporter plasmids, the corresponding regions of the human SIAT4A and GCP60 promoters were inserted into pGL4-basic vector (Promega). Isolated GASE sequences of target genes or 4×GASE from the human SIAT4A gene were inserted into the XhoI-BglII site of pGL3-promoter vector (Promega). Luciferase assays were carried out as previously reported, using Renilla luciferase as a standard for transfection eficiency (Yoshida et al., 2001b). A vector expressing YFP-Golgi was purchased from Clontech.
Immunoblotting and Northern blot analysis were carried out as described previously (Yoshida et al., 2001a). In vitro translation of ATF6 and XBP1 was carried out according to the manufacturer’s instructions (Promega). Anti-ATF6 and anti-XBP1 antisera were prepared in the laboratory of Dr. Kazutoshi Mori.
To induce Golgi stress, the capacity of the Golgi apparatus has to be made insufficient as compared with cellular demands. In the case of the ER stress response, inhibitors of ER function such as thapsigargin, tunicamycin, and dithiothreitol are used to induce ER stress (Yoshida, 2007b). Thapsigargin inhibits Ca++-ATPase, which is indispensable for maintaining a high concentration of Ca++ in the ER lumen (Price et al., 1992). Since ER chaperones, including BiP, require a high concentration of Ca++, the Ca++ leak caused by thapsigargin leads to inactivation of ER chaperones and then insufficiency of the ER function. We supposed that inhibitors of Golgi function, such as monensin and nigericin, could be utilized similarly to induce Golgi stress. Monensin is a carboxylic ionophore that binds Na+/H+, becomes incorporated into cholesterol-rich membranes and causes leakage of H+ from acidic organelles, including the Golgi apparatus, endosomes and lysosomes (Dinter and Berger, 1998). Since proteins located in these compartments require acidic environments, their functions are inhibited by the monensin-induced H+ leak. For instance, upon monensin treatment, post-Golgi transport of secretory proteins and lipids is inhibited, and modification of sugar chains in the Golgi apparatus, such as incorporation of galactose, sialic acid and fucose, is dramatically blocked (Tartakoff, 1983). Monensin also causes morphological changes of the Golgi apparatus such as swelling of Golgi cisternae to an extent easily visible at the light microscopic level (Dinter and Berger, 1998).
To check whether monensin was effective in the cells used in this study, HeLa cells expressing YFP fused with the Golgi-targeting signal derived from human beta 1,4-galactosyltransferase (the membrane-anchoring signal that targets the fusion protein to the trans-medial region of the Golgi apparatus) were treated with monensin (Fig. 1). In the absence of monensin, the Golgi apparatus was detected near the nucleus by fluorescent microscopy (Panel B), but was not visible by phase contrast microscopy (Panels A and C). Upon monensin treatment, swollen vesicles emerged near the nucleus (Panels D and G), and YFP-Golgi marker was located in the membrane of these vesicles (Panels E, F, H and I), which is consistent with previous studies (Dinter and Berger, 1998; Tartakoff, 1983). This suggests that most of these swollen vesicles were derived from the Golgi apparatus, and that monensin is effective in HeLa cells, in accord with the findings of Lamb and colleagues that monensin treatment inhibits post-Golgi transport of viral HA protein in HeLa cells (Sakaguchi et al., 1996). After 12 h of monensin treatment, the cytosol of HeLa cells was filled with numerous swollen vesicles (Panel J) in which YFP-Golgi was localized (Panels K and L). Most of these swollen vesicles did not seem to be autophagosomes or lysosomes, since autophagosomes emerge not near the nucleus but throughout the cytosol (Kuma et al., 2004), and are not large enough to be observed by phase contrast microscopy, and since the number and volume of lysosomes are not so markedly increased upon monensin treatment (Grinde, 1983).
 View Details | Fig. 1. Morphological change of HeLa cells induced by monensin or nigericin treatment. HeLa cells transiently transfected with a plasmid expressing YFP-Golgi marker were untreated (A–C), or treated with 0.3 μM monensin for 90 min (D-I) or 12 h (J–L), or with 1 μM nigericin (M–O) for 12 h. Cells were examined by phase contrast or fluorescence microscopy and photographed as indicated. Bars, 5 μm. |
To identify genes whose transcription is enhanced in response to disruption of Golgi function, RNA extracted from HeLa cells with or without monensin treatment was subjected to microarray analysis using arrays of a human whole cDNA library. Expression of 277 genes was found to increase by more than 2.0-fold upon monensin treatment. Among them, 26 were genes of unidentified function, while 32 genes were related to lipid synthesis and 5 were regulators of apoptosis, suggesting that one of the branches of the Golgi stress response might upregulate lipid synthesis for membrane expansion of the Golgi apparatus (such as observed in Fig. 1), and another branch might induce apoptosis. Interestingly, 10 genes had functions closely related to the Golgi apparatus (Table I). These Golgi-related genes encode proteins required for structural maintenance of the Golgi apparatus, glycosylation in the Golgi compartment, and post-Golgi vesicular transport, suggesting that another branch of the Golgi stress response acts to increase the capacity of the Golgi apparatus. GCP60 (Golgi complex-associated protein of 60 kDa) is a protein that binds to the Golgi-localization signal of giantin and is localized in the Golgi complex, and is involved in the maintenance of the Golgi structure (Sbodio et al., 2006; Sbodio and Machamer, 2007; Sohda et al., 2001). Overexpression of GCP60 results in mislocalization of GCP60, giantin and N-acetylglucosaminyltransferase I, and disruption of the Golgi structure. GCP60 contains a proline-rich domain that can bind the WW domain, which is found in a number of cytoskeletal proteins. Since GCP60 has no transmembrane domains, it may bind to a Golgi membrane protein to localize in the Golgi membrane. SIAT4A and SIAT10 are sialyltransferases localized in the trans-Golgi compartment (Berger and Roth, 1997; Kitagawa and Paulson, 1994), whereas FUT1 is a fucosyltransferase located in the trans cisternae of the Golgi apparatus. B3GAT2 is a glucuronyltransferase which is localized in the Golgi. UAP1L1 is a cytosolic UDP-N-acetylglucosamine pyrophosphorylase beta-like protein, and is involved in the biosynthesis of nucleotide-sugars, which are transported to the ER and Golgi, and used for glycosylation. STX3A (syntaxin3A) is a t-SNARE molecule located in compartments from the Golgi apparatus to the plasma membrane (Band and Kuismanen, 2005), whereas WIPI49 (a WD40 repeat protein of 49 kDa that interacts with phosphoinositides) is a protein involved in post-Golgi vesicular transport, and is localized in the trans-Golgi and endosomal membranes (Jeffries et al., 2004). RAB20 is a small GTPase involved in vesicular transport, while PCSK1 is a preprotein convertase that processes secretory proteins in the Golgi apparatus. We speculated that the expression of additional Golgi-related genes may be induced upon monensin treatment but our microarray analysis could not detect them because of their low abundance.
To precisely examine the time course of the induction of these candidate genes in response to Golgi stress, RNA extracted from HeLa cells treated with monensin or thapsigargin was subjected to Northern blot analysis (Fig. 2). SIAT4A, GCP60 and STX3A mRNAs were hardly increased by thapsigargin treatment (Panels A–C, lanes 1–6) but were remarkably upregulated by monensin treatment (lanes 7–12), whereas BiP mRNA was increased only by thapsigargin treatment, suggesting that the expression of SIAT4A, GCP60 and STX3A is specifically enhanced by Golgi stress, and that the mechanism of the Golgi stress response is completely distinct from that of the ER stress response. To confirm that monensin treatment does not activate the ER stress response, we examined the activation status of ATF6 or XBP1, key transcription factors regulating ER stress responses (Yoshida et al., 1998; Yoshida et al., 2001a) by immunoblotting. In the absence of thapsigargin, pATF6(P) and pXBP1(U) were observed (Fig. 3A and 3B, lane 3), whereas pATF6(N) and pXBP1(S) were emerged upon thapsigargin treatment (lane 4–7), indicating activation of the ER stress response by thapsigargin treatment. When cells were treated with monensin, pATF6(N) and pXBP1(S) were hardly detected (lanes 8–12), indicating that monensin hardly activates the ER stress response.
 View Details | Fig. 2. Northern blot analysis of Golgi-related genes during the ER and Golgi stress responses. Total RNA extracted from HeLa cells treated with 1 μM thapsigargin or 10 μM monensin for the indicated times was subjected to Northern blot analysis with (A) SIAT4A, (B) GCP60 and (C) STX3A probes. Each blot was also probed with BiP and GAPDH probes. RNA used for each blot was derived from the same experiment. |
 View Details | Fig. 3. Effect of monensin on ER stress markers. Whole cell lysates prepared from HeLa cells treated with 1 μM thapsigargin or 10 μM monensin were subjected to immunoblotting, and probed with (A) anti-ATF6α or (B) anti-XBP1 antiserum. Lanes 1 and 2 included in vitro translated proteins as indicated. Asterisks indicate nonspecific bands. |
We also tested the effect of nigericin, another Golgi-disturbing agent. Nigericin is a K+/H+ ionophore, which depletes H+ from the Golgi lumen similarly to monensin (Dinter and Berger, 1998). Upon nigericin treatment, the Golgi apparatus was expanded in HeLa cells (Fig. 1, panels M–O), as observed upon monensin treatment (panels J–L). Northern blot analysis revealed that expression of GCP60 mRNA was upregulated in response to nigericin treatment (Fig. 4, lanes 7–12) as well as monensin treatment (lanes 1–6). These results suggest that HeLa cells have a mechanism of Golgi stress response that compensates for loss of Golgi function by upregulating the expression of Golgi-related genes, and that monensin activates at least a part of the signaling pathways of the Golgi stress response.
 View Details | Fig. 4. Effect of nigericin on GCP60 expression. Northern blot analysis of GCP60 mRNA was carried out with RNA prepared from HeLa cells treated with 10 μM monensin or 1 μM nigericin as described for Fig. 2. |
To identify a cis-acting element regulating transcriptional induction by Golgi stress, we analyzed the promoters of the human SIAT4A and GCP60 genes. The promoter region encompassing –2,651 nt from the transcription start site of the human SIAT4A gene was fused with a firefly luciferase gene, and transfected into HeLa cells (Fig. 5A). Transcriptional induction by monensin was evaluated by measuring luciferase activity corrected by Renilla luciferase activity used as a standard of transfection efficiency. The full-length SIAT4A promoter responded to monensin treatment, resulting in a 2-fold increase of transcription of the luciferase gene (lane 1). When deleted to –296 nt, the SIAT4A promoter was still able to respond to monensin (lanes 2 and 3), whereas deletion to –271 abolished the induction by monensin (lane 4), suggesting that the [–296 to –271] region contains an enhancer element.
 View Details | Fig. 5. Promoter analysis of target genes of the Golgi stress response. (A and B) Deletion analysis of the SIAT4A and GCP60 promoters. HeLa cells transiently transfected with the indicated reporter constructs harboring human SIAT4A and GCP60 promoters and their deletion mutants were treated with or without 0.3 μM monensin, and the increase of luciferase activity upon monensin treatment was plotted. A reporter plasmid containing the SV40 promoter was used as a negative control. Error bars show the standard deviation from three independent experiments. (C) (upper panel) GASE-like sequences found in human SIAT4A, GCP60 and UAP1L1 promoters. (lower panel) Enhancer activity of each GASE-like sequence. Reporter plasmids containing the GASE-like sequences shown in the upper panel were transiently transfected into cells, and increase of luciferase activity upon monensin treatment was plotted. Mutants of each GASE-like sequence, in which the consensus sequence of GASE was disrupted as shown in the upper panel, were also included (indicated by Xes). (D and E) Effect of GASE elimination on the activity of SIAT4A and GCP60 promoters. Activity of human SIAT4A, GCP60 and their mutant promoters in which a GASE-like sequence was changed to a mutant sequence (from ACGTGGC to CATGTGC; indicated by Xes) were evaluated as described in (A). (F) GASE-like sequences found in the promoters of predicted target genes of the Golgi stress response. The promoter regions of genes listed in Table I were searched for sequences similar to ACGTGGC. Nucleotides that do not match the GASE consensus are shown in lowercase letters. Numbers show the position from a transcription start site. |
We performed a similar promoter analysis of the human GC60 gene, and found that the [–98 to +16] region contains an enhancer element responding to monensin treatment (Fig. 5B). The [–296 to –271] region of the SIAT4A promoter and the [–98 to +16] region of the GCP60 promoter contain a common sequence motif of ACGTGGC (Fig. 5C, upper panel). When isolated and joined with the luciferase gene, these short sequences containing ACGTGGC were able to respond to monensin treatment in HeLa cells (Fig. 5C, lower panel, lanes 1 and 3). If the ACGTGGC motifs were disrupted, these sequences did not enhance transcription upon monensin treatment (lane 2 and 4). Moreover, if the ACGTGGC motifs were disrupted in the long promoters of the SIAT4A or GCP60 gene, these promoters lost the ability to respond to monensin (Fig. 5D and 5E). These results suggest that transcriptional induction of the SIAT4A and GCP60 genes is commonly regulated by sequences containing an ACGTGGC motif. We named this novel cis-acting element the Golgi apparatus stress response element (GASE).
To identify the nucleotides important for transcriptional induction mediated through GASE, point mutations were systematically introduced into the GASEs of the human SIAT4A and GCP60 genes (Fig. 6). A point mutation of the first guanine to thymine did not affect the enhancer activity of SIAT4A-GASE (Fig. 6A, lane 3), whereas the mutation of any nucleotide in the ACGTGGC sequence abolished transcriptional induction (lanes 13–19), suggesting that the sequence of ACGTGGC is indispensable for the enhancer activity. In the case of GCP60, any point mutation of the first 5 nucleotides of ACGTGGC abolished transcriptional induction (Fig. 6B, lanes 13–17). As for the last two nucleotides, mutation of G to T (lane 18) or C to A (lane 19) hardly affected the activity, whereas mutation of G to A or C (lanes 23 and 24) or C to G (lane 25) abolished it. These findings suggest that the essential nucleotide sequence of the SIAT4A and GCP60 GASEs is ACGTGgt (the last two letters are in lower case letters since they are less required for the response).
 View Details | Fig. 6. Mutational scanning of GASE. A series of point mutants of (A) human SIAT4A and (B) GCP60 GASE were fused with the luciferase gene and transfected into HeLa cells, which were untreated or treated with 0.3 μM monensin. Lowercase letters indicate each point mutation. Enhancer activity of each construct was evaluated as in Fig. 5. |
GASE-like sequences were ubiquitously found in the promoters of target genes of the Golgi stress response (Fig. 5F). Indeed, a GASE-like sequence found in the promoter of the human UDP-N-acetylglucosamine pyrophosphorylase (UDP1L1) gene showed enhancer activity (Fig. 5C, lanes 6), and mutation of the ACGTGGC motif abolished it (lane 7). From these results, we concluded that transcriptional induction of Golgi-related genes in response to monensin treatment is commonly mediated by GASE.
To confirm that GASE regulates transcriptional induction in response to Golgi stress, we examined whether GASE-mediated transcription is induced when the function of the Golgi apparatus is disrupted by overexpression of GCP60, one of the target genes of the Golgi stress response identified here. Misumi and colleagues reported that GCP60 binds to the Golgi-localization signal of giantin, and that overexpression of GCP60 resulted in mislocalization of GCP60 and giantin to the cytosol, leading to fragmentation and dysfunction of the Golgi apparatus (Sohda et al., 2001) (Fig. 7A). When a reporter construct harboring four tandem repeats of SIAT4A-GASE fused with a luciferase gene was transfected into cells, overexpression of GCP60 significantly increased the luciferase activity (Fig. 7B lane 2), as compared with the vector control (lane 1), strongly suggesting that the signal activating transcription through GASE originates from the dysfunctional Golgi apparatus.
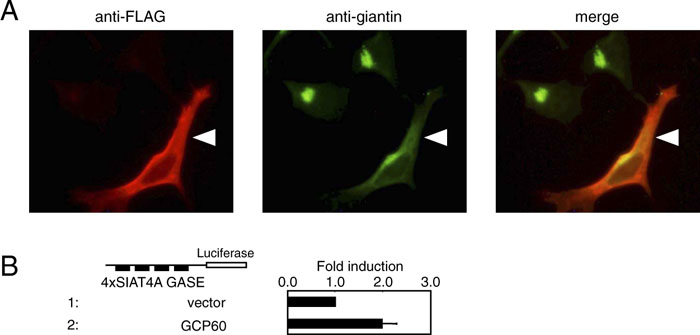 View Details | Fig. 7. Golgi stress caused by GCP60 overexpression induced GASE-mediated transcription. (A) Mislocalization of giantin by GCP60 overexpression. HeLa cells transfected with a FLAG-GCP60 expression plasmid were stained with anti-FLAG and anti-giantin antisera. A cell overexpressing GCP60 is indicated with an arrowhead. (B) Activation of GASE-mediated transcription by GCP60 overexpression. A reporter plasmid harboring four copies of GASEs of human SIAT4A gene was transiently transfected into HeLa cells together with a vector alone or a plasmid overexpressing human GCP60. GASE-mediated transcriptional activity was evaluated as described in Fig. 5. |
As the capacity of the ER is regulated by the ER stress response in accordance with cellular demands, we speculated that the capacity of the Golgi apparatus should be regulated by a similar regulatory system called the Golgi stress response, though the existence of the Golgi stress response has not hitherto been demonstrated and the molecular mechanism by which the Golgi stress response might occur remained unclear. Here we revealed that transcription of Golgi-related genes is induced when Golgi function is disrupted by monensin, nigericin, or GCP60 overexpression (Golgi stress), and that the transcriptional induction in response to Golgi stress is commonly regulated by a novel cis-acting element, GASE. These observations support the notion that mammalian cells have a mechanism of Golgi stress response to maintain the homeostasis of the Golgi apparatus.
In this study, monensin was used to induce Golgi stress, though this chemical might have unexpected pleiotropic effects on cells. We think that monensin can be utilized as a Golgi stress-inducing agent, for the following reasons. First, monensin treatment of mammalian cells actually enhances transcription of not a few Golgi-related genes (Table I), and transcriptional induction of these genes seems to be regulated by a common regulatory mechanism through GASE, suggesting the existence of a novel response system regulating expression of the Golgi-related genes. Second, we observed that nigericin induced transcription of GCP60 mRNA (Fig. 4), suggesting that neutralization of acidic organelles including the Golgi as well as lysosomes is the root cause of transcriptional induction by monensin. Third, only a few genes involved in lysosomal function were found to be induced by monensin treatment (Table II), suggesting that monensin only partially activates the lysosome stress response (see below). Finally, GASE-mediated transcription was actually induced when the function of the Golgi apparatus was impaired by overexpression of GCP60 (Fig. 7B). We think that GASE-mediated transcription of Golgi-related genes is induced by the Golgi stress response, whereas it is possible that transcription of non-Golgi genes mediated by cis-acting elements other than GASE might be upregulated by side effects of monensin, independent of Golgi stress response. This seems to be similar to the situation when thapsigargin is used as an ER stress inducer, in which an increase of cytoplasmic Ca++ enhances transcription of genes in an ER stress-independent manner. Further analysis focusing on GASE-mediated transcriptional induction may strongly facilitate breakthroughs, since there are few alternative methodologies to artificially activate Golgi stress response in experimental systems.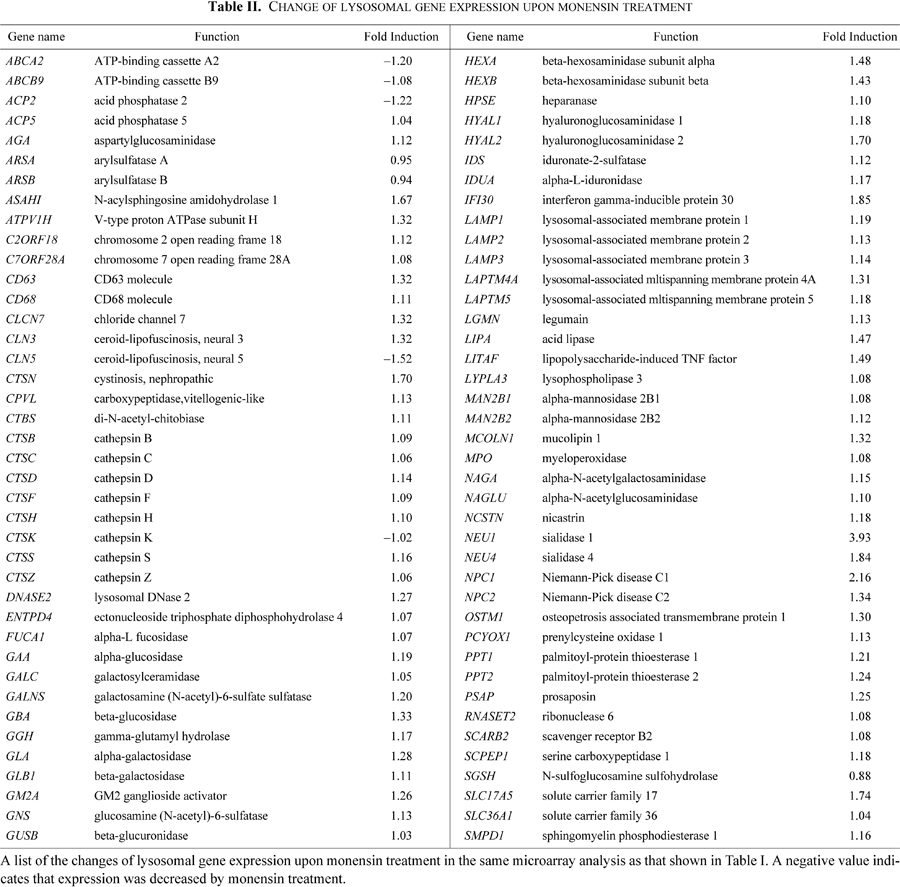
It is possible that monensin could neutralize the lysosome and disrupt its function, leading to a “lysosome stress response” (Sardiello et al., 2009). However, our microarray analysis showed that this was not the case (Table II). Expression of most lysosomal genes such as CTSD and CTSF, whose expression is commonly upregulated by the transcription factor TFEB and the cis-acting element CLEAR in response to lysosomal stress, was not induced upon monensin treatment in our microarray analysis, and expression of only two genes (NEU1 and NPC1) out of 80 lysosomal genes was upregulated more than 2-fold. We speculate that there are several reasons why monensin did not activate a lysosomal stress response. First, since monensin prevents secretory proteins from being transported to the lysosome in monensin-treated cells, few secretory proteins are accumulated in the lysosome. Thus, though monensin disrupts the activity of lysosomal enzymes, the capacity of the lysosome is not overwhelmed by unprocessed secretory proteins and the lysosome stress response is not activated. This would be a similar situation to the lack of ER stress response induction by ER stress inducers if the production of secretory proteins is blocked by cycloheximide treatment. Second, since monensin tends to be inserted into the cholesterol-rich membranes (Bransburg-Zabary et al., 1996) and cholesterol is accumulated in the trans-Golgi cisternae, monensin may have a stronger impact on the trans-Golgi compartment than on lysosomes (Dinter and Berger, 1998). Actually, when cholesterol localization was probed using filipin, a fluorescent chemical that specifically binds to cholesterol, the Golgi apparatus was found to be one of the major compartments rich in cholesterol in HeLa cells [Supplementary Fig. 1A]. Interestingly, swollen vesicles formed after monensin treatment were found to be remarkably rich in cholesterol [Supplementary Fig. 1B].
We named the response we observed here “the Golgi stress response” rather than an alternative such as “the Golgi disruption response”, because the term “stress response” is used not only for various situations in which the accumulation of unfolded proteins induces transcription of target genes, for instance, the ER stress response, but also for certain situations that are independent of the accumulation of unfolded proteins, such as the oxidative stress response and osmotic stress response. Moreover, it was recently reported that the capacity of the lysosomes is regulated by the mechanism of the “lysosomal stress response”, suggesting that each organelle has a mechanism regulating its capacity according to cellular needs (see below). Therefore, from the point of view of the regulatory mechanisms of organelle capacity, we think that the name “the Golgi stress response” is appropriate.
Recent progress of research on organelle biogenesis suggests that each organelle has an autoregulatory mechanism that adjusts its capacity in response to cellular demand (Yoshida et al., 2009). The capacity of the ER seems to be adjusted to cellular needs by the ER stress response or unfolded protein response, which regulates the expression of ER chaperones and protein degradation machinery (see Introduction). In the case of the mitochondria, transcription of genes encoding the mitochondrial chaperones and protein degradation machinery is activated by the mechanism called the mitochondrial unfolded protein response when unfolded proteins are accumulated in the matrix of the mitochondria. This transcriptional induction is regulated by the sensor molecule CLPP-1 and transcription factors DVE-1 and ZC376.7 in C. elegans (Haynes et al., 2007, 2010), and by the transcription factor CHOP in mammals (Ryan and Hoogenraad, 2007). The capacity to process fatty acids by β-oxidation in yeast peroxisomes is regulated by Zinc-finger type transcription factors Oaf1p and Pip2p (Karpichev et al., 1997). The Oaf1-Pip2 system is well conserved in mammals, and is regulated by transcription factors PPARα and RXR (Phelps et al., 2006; Reddy and Hashimoto, 2001). These transcription factors bind directly to and are activated by long chain fatty acids (Chakravarthy et al., 2009). Recently, it was reported that most lysosomal genes are regulated by the transcription factor TFEB (Sardiello et al., 2009). Under aberrant lysosomal storage conditions (lysosomal stress), TFEB translocates from the cytoplasm to the nucleus, resulting in the activation of lysosomal genes through a cis-acting element called CLEAR, whose consensus sequence is GTCACGTGAC.
In conclusion, here we reported evidence supporting the notion that mammalian cells have the Golgi stress response. As far as we know, this is the first report of the Golgi stress response. We delineated here only a part of the Golgi stress response, and further studies will be required to identify sensor molecules detecting Golgi stress, the sensing mechanisms, the molecular basis of Golgi stress, and the biological significance of the Golgi stress response. Clarification of the regulatory mechanism will contribute not only to establishing the new research field of organelle autoregulation, which addresses a fundamental problem in cell biology, but also to developing therapeutic agents for Golgi-related diseases (Fujita et al., 2006; Hu et al., 2007; Huynh et al., 2003; Jellinger, 2009; Zeevaert et al., 2008), in the same way that elucidation of the mechanism of the ER stress response has enhanced research regarding neurodegenerative diseases, including Alzheimer’s and Parkinson’s diseases.
We thank Dr. Kazutoshi Mori for providing the yeast one hybrid system and anti-ATF6α antiserum. We also thank Dr. Elizabeth Nakajima for critical reading of the manuscript, and Ms. Kaoru Miyagawa for technical-secretarial assistance. This work was supported by The Uehara Memorial Foundation, Sankyo Foundation of Life Science, the PRESTO-SORST program of the Japan Science and Technology Agency and grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (Nos. 21113511, 22370069 and 22020019). It was also financially supported in part by the Global Center of Excellence Program A06 “Formation of a Strategic Base for Biodiversity and Evolutionary Research: from Genome to Ecosystem” of MEXT and by a Hyogo Prefecture Government Grant.
|