| To whom correspondence should be addressed: Fumio Imamoto, Department of Molecular Biology, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan. Tel: +81–6–6879–8325, Fax: +81–6–6879–8335 Present address: Takefumi Sone, Division of Gene Therapy, Research Center for Genomic Medicine, Saitama Medical University, 1397-1 Yamane, Hidaka, Saitama 350-1241, Japan. Present address: Taichi Andoh, Life Technologies Japan Ltd., 10-28 Hirochiba-cho, Suita, Osaka 564-0052, Japan. |
Induced pluripotent stem cells (iPSC) are generated from somatic cells by transduction of four transcription factors, Oct4, Sox2, Klf4, and c-Myc (Takahashi and Yamanaka, 2006). An important consideration in the design of multi-gene delivery technology is the availability of a suitable vector to stably and stoichiometrically introduce multiple genes into living cells and co-express these genes efficiently. As a promising system for this purpose, we have developed all-in-one expression constructs harboring tandemly situated multiple cDNAs and their expression control signals in a single vector. One improvement to the current strategy would be the delivery of multiple reprogramming factors within the context of a single vector. Construction of multiple functional DNA elements in tandem on a single plasmid has been performed by stepwise Gateway recombination reactions using Multisite Gateway technology (Life Technologies, Carlsbad, USA) (Cheo et al., 2004; Inoue et al., 2009; Sasaki et al., 2004; Sone and Imamoto, 2011; Sone et al., 2008).
Although the expression from tandemly situated multiple genes with independent promoters often suffers from transcriptional interference, insertion of insulator sequences such as chicken HS4 can markedly alleviate it (Yahata et al., 2007). Using autonomous self-cleaving 2A peptides is also a method to avoid the transcriptional interference and achieve stoichiometric expression of multiple genes from a single promoter (Szymczak et al., 2004). Although polycistronic vectors with 2A-linked reprogramming factors have been widely used in generating iPS cells both in viral and non-viral methods (Carey et al., 2009; Kaji et al., 2009; Sommer et al., 2009; Woltjen et al., 2009; Ye et al., 2010), non-integrating methods for generating iPSC using plasmid, oriP/EBNA-1 (Epstein-Barr nuclear antigen-1)-based episomal and ‘minicircle’ vectors (Okita et al., 2008; Yu et al., 2009; Jia et al., 2010), still have generally suffered from low reprogramming efficiencies, mostly due to low transduction rates against the primary somatic cells. Reprogramming by adenoviral vectors has been expected to overcome the problem of low transduction efficiencies of non-integrating vectors, the efficiency of reprogramming was, however, still low (Stadtfeld et al., 2008; Zhou and Freed, 2009).
A typical 4 genes-in-one construct used for iPSC generation in the present experiments is about 12.6 kb in molecular size (Fig. 1b or c). The utility of such a relatively large expression plasmid is limited by loading capacity of the transduction system into live cells; i.e., the retroviral and lentiviral vectors can pack the foreign DNA only in less-sized molecule than about 5–7 kb without considerable reduction of titer (Park, 2007), however in contrast to this, the BacMam transduction system has a large payload capacity up to 38 kb or more (Cheshenko et al., 2001).
 View Details | Fig. 1. Diagrammatic maps of BacMam bacmid and oriP/EBNA-1 plasmid clones used. Schematic maps of BacMam bacmid (a–g) and oriP/EBNA-1 plasmid clones (h–k). (a) Venus-reporter (bCAG/BV2-Venus), (b) OKSM-2A2 (bCAG/BV2-hOKSMiG), (c) OSKM-2A2 (bCAG/BV2-hOSKMiG), (d) EOS-reporter (bBV2-EOS-S(4+)-EmGFP), (e) OKSM-2A3 (bCAG/BV2/EBNA-OKSMiG), (f) OSKM-2A3 (bCAG/BV2/EBNA-OSKMiG), (g) LT-2A2 (bCAG/BV2-SV40LT), (h) tandem O-S (pCAG/EBNA-mO-mS), (i) tandem K-M (pCAG/EBNA-mK-mM), (j) LT (pCAG/EBNA-SV40LT) and (k) polycistronic OKSM (pCAG/EBNA-mOKSM). The full names of BacMam bacmid clones are correspond to the names of the plasmid clones shown in Table IV when the initial “b” is replaced with “p”. The expression cassettes, which are flanked by Tn7R and Tn7L, are common between the plasmid and bacmid clones (a–g). The expression of Oct4, Klf4, Sox2, c-Myc, Venus-reporter and SV40LT is directed by respective CAG promoter (pCAG) (a–c, e–f, h–k) except that EmGFP is directed by EOS-S(4+) promoter (pEOS) for EOS-reporter (d). In two tandem O-S and K-M constructs (h, i), a cHS4 insulator (filled box) is inserted between the transcription units. Closed circles represent poly-adenylation (pA) signals of SV40 or bovine growth hormone (BGH). Wavy lines represent the bacmid backbone of bMON14272. 2A indicates FMDV 2A peptide sequences. B1~B6 indicate attB signals, remnants of Multisite Gateway cloning. GmR indicates a gentamycin resistance gene. HygR indicates a hygromycin resistance gene. |
Recombinant baculoviruses engineered to contain a mammalian expression cassette have been shown to efficiently transduce mammalian cells (Boyce and Bucher, 1996; Hofmann et al., 1995). These viral vectors containing a mammalian promoter upstream of the target genes were termed BacMam viruses. This system has evolved rapidly over the last several years (Kost et al., 2005), for example, for rapid and high-level protein production in mammalian cells for the purpose of drug screening (Kost et al., 2007), multiple transporters studies (Dukkipati et al., 2008) and protein structural studies (Dukkipati et al., 2008). Further applications of the baculovirus system have expanded to eukaryotic gene display, cancer therapy, tissue engineering and a potential vaccine delivery, (Hu, 2008).
The BacMam system is based on site-specific transposition of an expression cassette into a baculovirus shuttle vector (bacmid) propagated in E. coli (Ciccarone et al., 1997; Luckow et al., 1993). One major advantage of the BacMam system is that efficient transduction is achieved in many different mammalian cell types by simply adding a viral inoculum (Condreay et al., 1999). In addition, several parameters have been explored to improve transduction of those cell types which are not efficiently transduced (Kost et al., 2005). The BacMam Gateway system has been developed to construct an engineered viral genome, a bacmid, containing the gene(s) of interest and viral backbone elements, vesicular somatitis virus G protein (VSV-G) (Barsoum et al., 1997) and woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) (Deglon et al., 2000), which enhances transduction efficiency and increases persistence of expression, respectively (Life Technologies, Carlsbad, USA). Another advantage of this system as an efficient gene delivery tool is to be able to accommodate large insertions of foreign DNA, initiating little to no cytopathic effect in mammalian cells and hence have a good biosafety profile (Kost and Condreay, 2002).
We report here that the BacMam transduction system combined with a multi-gene expression vector could lead to efficient transduction and successful generation of iPS cells, thus presumably achieving an effective stoichiometric balance of four factors in the cells which is assumed to be important to generate fully reprogrammed iPS cells (Yamanaka, 2009a).
For investigating transduction efficiency of BacMam particles with primary mouse embryonic fibroblast (MEF) cells, we constructed a reporter gene expression plasmid carrying a Venus gene as indicated in Fig. 1a. BacMam particles containing this reporter plasmid were prepared and used in a triple (successive) transduction protocol into MEF cells at intervals of 3 days. The transduction efficiency, relative number of transductants per total cells, determined by FACS assay was 88–97% one to two days after the 1st transduction, and 64–98% at the eighth day after the third transduction. The BacMam-Venus reporter plasmid construct with version 2-backbone (Life Technologies, Carlsbad, USA) which contains VSV-G and WPRE for enhancing transduction efficiency and increasing persistence of expression, respectively, was more efficiently transduced into MEF cells (data not shown). Viability of MEF cells was not noticeably affected during transduction and cultivation thereafter with the BacMam particles.
Generation of iPS cells from mouse embryonic fibroblast (MEF) was achieved by BacMam-transduction of a polycistronic plasmid expression vector for coincident and optimized expression of four defined reprogramming transcription factors, Oct4, Klf4, Sox2 and c-Myc in this order on a single plasmid. A four-in-one expression plasmid for bacmid (OKSM-2A2, outlined in Fig. 1b) was constructed by joining 3 DNA fragments using Multisite Gateway technology. The plasmid backbone of this construct included VSV-G and WPRE sequences for broad host range and increased expression respectively. The 4 cDNAs were fused in a single ORF via self-cleaving 2A peptides (Szymczak et al., 2004). In order to compare the efficiency for generating iPS cells of the Oct4-Klf4-Sox2-c-Myc (OKSM-2A2) expression plasmid, the Oct4-Sox2-Klf4-c-Myc (OSKM-2A2) expression plasmid shown in Fig. 1c was also constructed. The expression of these ORFs was directed by the CAG promoter.
After 3 successive transductions of MEF cells (at intervals of 3 days) with BacMam particles carrying a OKSM-2A2 cassette, iPS cell colonies were observed in 15–24 days after the 1st transduction. With the iPS cell lines obtained from these colonies, the reprogramming state was investigated by immunofluorescence analyses with Nanog and SSEA-1, EOS-S(4+)-EmGFP reporter and alkaline phosphatase substrate staining. As indicated in Fig. 2, all of iPS cell lines tested were positively stained, thus exhibiting that these cell lines are in a reprogrammed state. To extend the analysis, iPS cell colonies formed at about 16 days after the 1st transduction were developed to regularly pulsating cardiomyocytes on the plate by 21 days after the 1st transduction (data not shown), thereby exhibiting their pluripotent activity. As a more stringent test of pluripotency, MEF-derived iPS cells obtained from transductants with OKSM-2A2 (Fig. 2) were injected into the testis of immunodeficient SCID mice to assay for teratoma formation. Tumors were recovered after 5–6 weeks. Histological analysis indicated the existence of tumors from all three germ layers, such as neural cells and epidermis (ectoderm), gut-like epithelium (endoderm) and muscle (mesoderm) (Fig. 3).
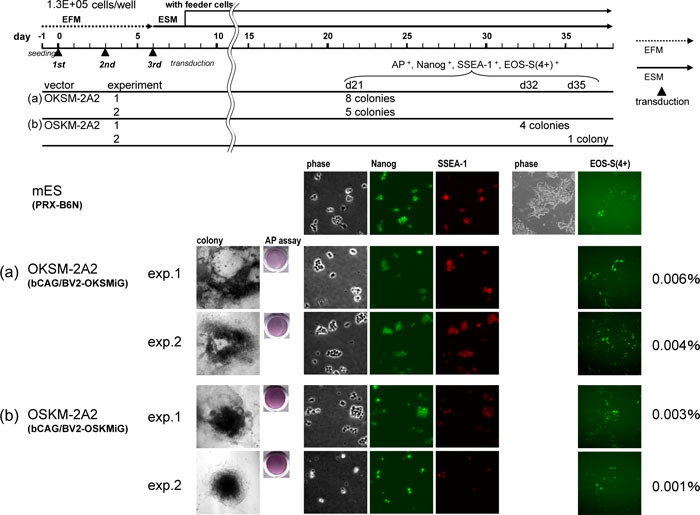 View Details | Fig. 2. Generation of iPSC using BacMam particles harboring OKSM and OSKM constructs. Successive triple transduction of MEF with BacMam particles of (a) OKSM-2A2 (Fig. 1b) and (b) OSKM-2A2 (Fig. 1c) at a cell density of 1.3×10e5 cells/well in a 6-well plate was performed and colonies of ES-like cells were isolated under timelines and conditions shown in upper diagram. Two independent experiments were repeated for each particle (exp.1 and exp.2). In the end, 8, 5, 4 and 1 lines were established as iPS cell lines for each experiment, (a) OKSM-2A2 exp.1 and exp.2, (b) OSKM-2A2 exp. 1 and exp.2, judging by morphology, AP assay, and immunofluorescence of Nanog and SSEA-1. All the lines were positive for EOS-S(4+)-EmGFP reporter assay. The efficiencies of iPSC generation from 1.3×105 MEF cells were calculated as 0.004–0.006% and 0.001–0.003% for OKSM-2A2 and OSKM-2A2, respectively, and are shown in the right end of the panels (a and b). The efficiency and the timing of colony formation were significantly better for OKSM-2A2 than OSKM-2A2. In the bottom panels from left, imaging profiles of isolated colony, AP assay, immunofluorescence data of phase contrast, Nanog and SSEA-1, and EOS-reporter assay data of phase contrast (not for all) and EmGFP fluorescence are shown for representative iPS cell lines derived from each experiment. The control imaging panels of a mouse ES cell line (PRX-B6N) are also shown above the iPS cell lines. |
 View Details | Fig. 3. Pluripotency of iPS cells derived from MEF cells. Hematoxyllin eosin staining of teratomas generated from MEF-derived iPS cells. Differentiated structures from all three germ layers including epidermis (a), epidermis (b), gut-like epithelium (c) and muscles (d) were found. Scale bars are 50 μm. |
Another construct containing reprogramming genes in an alternative order (OSKM-2A2) was repeatedly found to be less efficient in generating iPS cells than OKSM-2A2 construct. The smaller number of iPS cell colonies from OSKM-2A2 transductants appeared only after 32–35 days after the 1st transduction (Fig. 2b). However, the iPS cell lines from colonies picked up from OSKM-2A2 transductants could be sufficiently stained by Nanog and SSEA-1 immunofluorescence, EOS-S(4+)-EmGFP reporter and alkaline phosphatase substrate markers.
The efficiency of iPSC generation of polycistronic BacMam system was better than the polycistronic lentiviral system; i.e., in the present polycistronic BacMam system, the efficiency of iPSC production from 1.3×105 MEF cells was 0.004–0.006% and 0.001–0.003% for OKSM-2A2 and OSKM-2A2, respectively, as shown in Fig. 2, in contrast to the efficiency of 0.0001% of polycistronic (a similar construct to OSKM-2A2) lentiviral system (Carey et al., 2009).
Because the genome of baculovirus has been reported to not replicate in mammalian cells (Tjia et al., 1983), the baculoviral transduction vector may possibly be lost or diluted soon after transduction, resulting in short duration of transgene expression. Hence oriP/EBNA-1 elements have been used to construct baculovirus-based transduction vectors for mammalian cells for prolonged vector maintenance and enhanced transgene expression (Shan et al., 2006). Prolonged reporter gene expression has been reported with several cell lines, such as HEK293, Hone-1, Vero and Cos-7, although the duration of gene expression varied from 12 to 60 days in these different cell lines (Shan et al., 2006). We constructed baculoviral vectors containing oriP/EBNA-1 elements (Fig. 1e–g) and tested their efficiency in generating iPSC one month after the 1st transduction of MEF cells. As seen in Fig. 4a, both BacMam particles containing OKSM-2A2 or OKSM-2A3 generated essentially a comparable number of iPS cell colonies in 16-21 days after the 1st transduction. It has been reported that the inability of the baculoviral vectors to replicate in the mammalian cells is an advantage of BacMam transduction system in delivering the foreign genes without genomic integration (Merrihew et al., 2001; Zeng et al., 2009). However, as far as we determined preliminarily intracellular residual vectors in 4 iPS cell lines established by semi-quantitative genomic PCR method, the vector DNA could be detected in these cells (data not shown). Although this must be investigated further extensively with more cell lines, the possibility cannot be excluded that the efficiency of iPS cell derivation is significantly reduced without genomic integration (Okita et al., 2008; Stadtfeld et al. 2008). Two approaches have been known to remove transgenes from mouse or human cells, one is Cre/LoxP recombination system to excise integrated transgenes (Kaji et al., 2009; Soldner et al., 2009), and another is seamless excision of piggyBack transposons (Woltjen et al., 2009).
 View Details | Fig. 4. Generation of iPSC using BacMam particles harboring OKSM and OSKM constructs in presence of oriP/EBNA-1 element. The conditions used are essentially same as described in Fig. 2, except that the MEF cells were transduced 1 time (only 1st transduction) in (a) or 3 times in (b) with the multigene BacMam particles. In the end, 20, 15, 4, 0 and 20 lines were established as iPS cell lines for each experiment, (a) OKSM-2A2 (Fig. 1b), OKSM-2A3 (Fig. 1e), OSKM-2A3 (Fig. 1f), (b) OSKM-2A2 (Fig. 1b), judging by morphology, AP assay, and immunofluorescence of Nanog and SSEA-1. All the lines were positive for EOS-S(4+)-EmGFP reporter assay. The efficiencies of iPS cell generation calculated as described in Fig. 2 are shown in the right end the panels. In the bottom panels from left, imaging profiles of colony formation on plate, AP assay, immunofluorescence data of phase contrast, Nanog and SSEA-1, and EOS-S(4+)-EmGFP reporter assay data of phase contrast (not for all) and EmGFP fluorescence are shown for representative iPS cell lines derived from each experiment. The control imaging panels of a mouse feeder-free ES cell line (CGR8) are also shown above the iPS cell lines. |
It has been reported that the addition of baculovirus to mammalian cells in mid-log phase, followed by three further successive additions of virus (superinfection) at intervals of 3–4 days allows recombinant gene expression to be prolonged for over 10–15 days without addition any enhancing chemical such as butyrate (Hu et al., 2003). However, in our present transduction conditions with the multigene-BacMam vector carrying the VSV-G and WPRE elements, repetitive transduction was not always needed for efficient generation of iPS cells (i.e. a single transduction of MEF cells with baculoviral OKSM-2A2 particles seemed to be same or slightly more effective in inducing iPS cells after 16–25 days, Fig. 4b). This increased efficiency is compared with a three stage transduction where iPS cell colonies were generated at 21–25 days after the 1st transduction (Fig. 4a). These results suggest that the oriP/EBNA-1 elements are not required for increased efficiency in our present system. A possible explanation for this is that VSV-G and WPRE elements contained in the vector backbone function in increasing persistency of bacmid vectors in pluripotent cells.
The low efficiency and heterogeneous nature of reprogramming is a major impediment to the generation of useful iPS cell lines (Takahashi et al., 2007; Yamanaka, 2009a). Methods to identify the best mouse iPS cell clones from numerous candidates have included evaluation of cell morphologies, gene expression patterns and differentiation capacity, depending on the subsequent applications (Ellis et al., 2009; Maherali and Hochedlinger, 2008; Yamanaka, 2009b). Recently, an accurate pluripotency reporter system, which can be specifically mark pluripotent stem cells, has been developed. The EOS vector, carrying an early transposon promoter and Oct4 and/or Sox2 enhancer(s) can be used as an effective isolation of iPS cell lines that are expressing endogenous pluripotent markers (Hotta et al., 2009b). The EOS expression cassette regulates an enhanced green fluorescent protein (EGFP) gene downstream from the promoter. The EOS cassette is activated upon reprogramming in iPS cells, presumably by recruitment of the transcriptional activators to a mouse early transposon (ETn) promoter region and of the pluripotent state-specific transcriptional factors Oct4 and/or Sox2 to the enhancer region (Hotta et al., 2009b). Thus the EGFP reporter protein allows live-cell visualization in the process of reprogramming of iPS cells.
The conventional system for stable reporter gene transfer into primary somatic cells has been retroviral or lentiviral vectors, which may suffer from frequent transcriptional silencing in iPS cells (Okita et al., 2007; Wernig et al., 2007). Using the strong LTR promoter from ETn and Oct4 and/or Sox2 binding motifs, the EOS viral vector reporters robustly mark iPS cells with EGFP during reprogramming by overcoming vector silencing (Hotta et al., 2009a; Hotta et al., 2009b). We used EOS-S(4+)-EmGFP reporter (Hotta et al., 2009b) manufactured as BacMam viruses (Fig. 1d; kindly provided by Drs. Uma Lakshmipathy and Pauline Lieu of Life Technologies, R&D) in order to mark reprogrammed iPS cells.
We first investigated whether the EOS reporter can distinguish the heterogeneous nature of the iPS cell population, i.e., the reporter can exhibit its ability to discriminate a sufficiently reprogrammed iPS cells from partially reprogrammed cells. Electroporation has been used successfully to deliver the target DNA plasmid to a variety of cells (Potter and Heller, 2010). Since the electroporated DNA has inserted randomly into the genome of cells with a significant gene dosage dispersion in cell to cell, heterogeneous expression would be expected in the iPS cell population. This was demonstrated by using a mixture of equivalent amounts of two different types of tandem 2 cDNA expression constructs, murine Oct4-Sox2 (tandem O-S) and murine Klf4-cMyc (tandem K-M), directed by the CAG promoter (Fig. 1h, i). These constructs carry the oriP/EBNA-1 element for prolonged expression of reprogramming factors. In an attempt to counteract the possible toxic effects of reprogramming factors, we included an expression plasmid (Fig. 1j) of SV40LT (LT) in the experiment using electroporation (Hahn et al., 1999; Yu et al., 2009).
As shown in Fig. 5a, six iPS cell lines obtained from colonies generated at the 22nd day after electroporation were stained with immunofluorescence analyses with Nanog and SSEA-1 and alkaline phosphatase substrate staining assay. However, only one iPS cell line (#4–6) emerged with EmGFP expressed in transformed cells with EOS-S(4+)-EmGFP reporter BacMam particles, and the other five cell lines (including #4–33) lacked ability to express EOS-S(4+)-EmGFP reporter (Fig. 5c), indicating that these were not sufficiently reprogrammed even though Nanog, SSEA-1 and alkaline phophatase activities were exhibited. Results obtained from another series of similar experiments, carried out using a mixture of tandem O-S and K-M plasmids, was consistent to the above data; i.e., of six iPS cell lines isolated from colonies picked up at 22nd day after electroporation, only two cell lines emerged EmGFP positive paired with positive results with Nanog, SSEA-1 and alkaline phosphatase analyses. The other four cell lines did not express EmGFP from the EOS-S(4+) reporter although they exhibited positive results with Nanog and SSEA-1 staining analyses, and alkaline phosphatase activity (data not shown).
 View Details | Fig. 5. Heterogeneous nature of iPSC population generated by electroporated plasmid expression vectors. Transduction by electroporation were performed for two sets of oriP/EBNA-1 plasmid clones, (a) tandem O-S (Fig. 1h)+tandem K-M (Fig. 1i)+LT (Fig. 1j) and (b) polycistronic OKSM (Fig. 1k)+LT (Fig. 1j), and colonies of ES-like cells were isolated under timelines and conditions shown in upper diagram. It is slightly different from that of BacMam transduction for the starting point of using feeder cells and the use of ESM with conditioned medium (ES C.M.). In the end, 6 and 3 lines were established as iPS cell lines for each condition, (a) and (b), judging by morphology and AP assay. Then immunofluorescent analysis and EOS-reporter assay were performed for each cell line. The result are depicted by symbols of “+” or “–” under the middle panels of AP assay. Although all the lines were positive for Nanog and SSEA-1 (green “+”), 5 lines of (a) tandem O-S+tandem K-M+LT were negative for EOS-S(4+)-EmGFP reporter assay (black “–”). Cell lines of (a) tandem O-S+tandem K-M+LT starts with “#4-” and those of (b) polycistronic OKSM+LT starts with “#5-”. In bottle panels from left (c), imaging profiles of isolated colony, immunofluorescence data of phase contrast, Nanog and SSEA-1, and EOS-S(4+)-EmGFP reporter assay data of phase contrast and EmGFP fluorescence are shown for representative iPS cell lines of EOS-S(4+) positive (#4–6) and negative (#4–33). The control imaging panels of a mouse feeder-free ES cell line (CGR8) are also shown above the iPS cell lines. |
Insufficient and incomplete reprogramming processes have been discussed and successful reprogramming may depend on a specific stoichiometric balance of the four transcription factors (Oct4, Sox2, Klf4 and c-Myc, Yamanaka, 2009a). In so far as transduction systems employing baculoviral vectors and electroporated plasmids, an expression vector construct carrying four factor cDNAs (OKSM) on a single vector has been shown to be more efficient in generating iPS cells than a mixture of 2 tandem O-S and K-M expression vectors or a mixture of a single O, S, K and M expression vectors. As seen in Fig. 5b, the electroporated murine 4 cDNAs-in-1 vector (polycistronic OKSM) expression plasmid (Fig. 1k) generated iPSC colonies in 22 days after electroporation and all iPS cell lines from these colonies emerged with EmGFP expressed after application of the EOS-S(4+) reporter BacMam particles. On the other hand, a mixture of equivalent amounts of 4 single murine O, K, S and M expression plasmids failed to generate iPS cell colonies during incubation for 31 days after electroporation (data not shown). This is consistent with previous notion that efficiency of iPS cell generation was reduced when the four factors with separate viral vectors were introduced into cells, comparing with 2A-linked multiple cDNAs in one vector (Okita et al., 2008). A trial to introduce 4 genes by using a mixture of single gene carrying constructs would be challenging due to the inability to precisely control the transfection efficiency of each plasmid and the resulting potential variable gene dosage between cells.
As represented in Fig. 2 and Fig. 4, all of the iPS cell lines generated with BacMam transduction systems OKSM-2A2 and OSKM-2A2 effectively expressed EmGFP in cells transduced with EOS-S(4+)-EmGFP reporter viral particles. We have analyzed 38 iPS cell lines obtained so far, and confirmed that all of these iPS cell lines exhibited bright imaging profiles with EmGFP, as seen in those profiles represented in Fig. 2 and Fig. 4. In summary, the results presented here show the BacMam system as an efficient technology for transducing primary somatic cells to generate fully reprogrammed iPS cells in relatively short period without damaging cell physiology.
cDNAs of human and murine reprogramming factors (c-Myc, Oct4, Sox2 and Klf4) and SV40LT genes were amplified by normal or one-step adaptor PCR (Kagawa et al., 2004) using primers and templates listed in Table I. Template clones for human factors with ID (e.g. FLJ93327AAAN) are Gateway entry clones which were derived from the Human Genome and Protein Database (HGPD) (Goshima et al., 2008; Maruyama et al., 2009). Templates for murine factors (pMXs-Oct3/4, -Sox2, -Klf4, -c-Myc) were originally cloned in Yamanaka’s laboratory (Takahashi and Yamanaka, 2006) and obtained through Addgene. The PCR products were cloned into cloning vectors (pDONR-P1P4 (Sasaki et al., 2004) or pCR®2.1-TOPO® (Life Technologies, Carlsbad, USA) by the methods listed in Table I and confirmed by sequencing. Sequences of the cloning and sequencing primers are shown in Table II.

pCR2.1-(m)Oct4-2A, pCR2.1-(m)Sox2-2A or pCR2.1-(m)Klf4-2A, was digested with SalI and XbaI and the cDNA fragment with C-terminal foot-and-mouth disease virus (FMDV) 2A peptide was excised. Then each fragment was inserted between neighboring SalI and SpeI sites at N-terminal end of pENTR-L1-k-(m)cMyc-*L4. Resulting entry clones with tandemly linked cDNA by FMDV 2A peptide sequence also have neighboring SalI and SpeI sites at the N-terminus. Subsequently, another cDNA fragment with 2A peptide sequence digested with SalI and XbaI was inserted between the SalI and SpeI sites and this step was repeated until all 4 factors are linked with 2A peptide. Two sets of entry clones (OKSM and OSKM) of human and murine reprogramming factors were constructed in this manner and used in further experiments.
Four L1-L2 entry clones of murine reprogramming factors were obtained by BP reaction of the pMXs series and pDONR®201 (Life Technologies, Carlsbad, USA).
Another entry clone, pENTR-R4-pASV40-CV-PCAG-R3 with a cassette of SV40 pA signal (pASV40), five times repeat of chicken HS4 core insulator (CV) and CAG promoter (PCAG) was constructed by replacing BGH pA signal and EF-1α promoter of pENTR-R4-pABGH-PEF1α-R3 (Sone et al., 2008) with corresponding DNA fragments (191 bp and 1322 bp, respectively), which were excised from pENTR-R2-pASV40-L4 (Sone et al., 2009) and pENTR-L1-PCAG-R3 (Sone et al., 2009) with XbaI and HindIII, respectively. Then 5x cHS4 core fragment, which was excised from pUC-5xcHS4 core (Yahata et al., 2007), was inserted into unique SalI site of pENTR-R4-pASV40-PCAG-R3.
All the names of entry clones used in this paper are listed in Table III with their contents and origins.
Three BacMam (pBV1-DEST, pBV2-DEST and pBV2/EBNA-DEST) and one oriP/EBNA-1 (pEBNA-DEST) destination vectors were kindly provided by R&D of Life Technologies, Carlsbad, USA.
Modified versions of pBV2-DEST and pBV2/EBNA-DEST with a CAG promoter were constructed by inserting a CAG promoter fragment (1747 bp), which was excised from HindIII-digested pENTR-L5-PCAG-R3 (Inoue et al., 2009), into unique HindIII site of pBV2-DEST or pBV2/EBNA-DEST. A modified pEBNA-DEST with CAG promoter was constructed by replacing attR1-CmR-ccdB cassette (1614 bp) of SalI-digested pEBNA-DEST with PCAG-attR1-CmR-ccdB cassette (3367 bp), which was excised from SalI-digested pCAG/B2B1/SV-CV-DEST. pCAG/B2B1/SV-CV-DEST was constructed by inserting pASV40-CV cassette (1411 bp), which was excised from XbaI-digested pENTR-R2-pASV40-CV-L3 (Sone et al., 2009), into unique XbaI site of pEF/B2B1/V5-DEST, and then by replacing the EF-1α promoter (1322 bp) with the CAG promoter (1747 bp) of HindIII-digested pENTR-L5-PCAG-R3 (Inoue et al., 2009).
All the names of destination vectors used in this paper are listed in Table III with their special features and origins.
BacMam and oriP/EBNA-1 plasmid expression clones were constructed by Multisite Gateway LR reactions (Sasaki et al., 2004; Yahata et al., 2005). The entry clones and destination vectors used to construct each expression clone are listed in Table IV. The names of BacMam plasmid clones are correspond to the names of the bacmid clones shown in Fig. 1a-g when the first “p” is replaced with “b”. The expression cassettes, which are flanked by Tn7R and Tn7L, are common between the plasmid and bacmid clones.
For construction of complex expression clones such as tandem O-S or K-M, a stepwise Multisite Gateway method was performed as described (Inoue et al., 2009; Sone et al., 2009; Sone et al., 2008). Essentially, temporal expression clones were first constructed by LR reaction (Table IV, temporal pEXPR), then they were converted into modular entry clones by BP reaction (Table IV, modular pENTR).
Generation of recombinant BacMam bacmids and viral particles was performed essentially following the manual of the Bac-to-Bac® Baculovirus Expression System (Life Technologies, Carlsbad, USA). The purified BacMam expression clone plasmid DNA was used to transform DH10BacTM chemical competent cells (Life Technologies, Carlsbad, USA). The plasmid expression cassette, which is flanked by left and right arm Tn7 sequences, was transposed into a mini-attTn7 target site of the bacmid, bMON14272, contained in DH10Bac, catalyzed by Tn7 transposase encoded by a helper plasmid, pMON7124, also contained in DH10Bac (Luckow et al., 1993). The recombinant bacmid clones were identified as white colonies by blue-white selection because transposition into the mini-attTn7 target site disrupts the LacZα peptide in which the target site is situated. After purification of the bacmid DNA, PCR analysis using the primers M13-Rv and one of M13-Fw or Rv-7 CMV (Table II) was performed to verify the presence of the expression cassette in the recombinant bacmid.
Once the recombinant bacmids were obtained, they were transfected into SF9 cells (Life Technologies, Carlsbad, USA) by using Cellfectin® II Reagent (Life Technologies, Carlsbad, USA) to generate recombinant baculoviral particles (P1). P1 viral stock was obtained after incubation at 27°C for 72 hours. Titer of the P1 viral stock was determined by plaque assay and single plaques were purified (if necessary). The P1 stock or the plaque-purified stock was used for a second and third round of transfection into SF9 cells to generate P2 and P3 stocks. After the baculoviral stocks were amplified and the titer was determined, these high-titer stocks (~5×107 pfu/mL) were stored at 4°C in the dark.
Transduction of mouse embryonic fibroblast (MEF) cells with BacMam particles was performed essentially following the PremoTM Cameleon Calcium Sensor manual (Life Technologies, Carlsbad, USA). MEF prepared from E12.5 embryo of C57BL/6N strain were seeded in embryonic fibroblast media (EFM) {10% Fetal Bovine Serum (FBS), 0.1 mM (1x) MEM Non-Essential Amino Acids Solution (NEAA), 1x Penicillin-Streptomycin-Glutamine (PSQ), 100 μM 2-mercaptoethanol (2-ME) in IMDM (Iscove’s Modified Dulbecco’s Medium)} at 1.3×105 cells/well in 6-well plate one day before transduction. The amount of the materials was reduced (or increased) according the scale of the experiment. On the day of the 1st transduction, the medium was replaced with a mixture of 400 μL BacMam particles and 700 μL PBS medium then incubated for 4 hr at room temperature. Then the medium was removed and 2 mL EFM containing 2 μL of 1 mM trichostatin A (TSA) was added followed by incubation for 2 hr at 37°C, 5%CO2. Medium was changed to normal EFM and incubated for 3 days at 37°C, 5%CO2. The 2nd and the 3rd transductions were performed on day 3 and day 6, if necessary. The protocols were almost identical with the 1st transduction unless incubation time with BacMam particles were shortened from 4 hr to 2 hr and embryonic stem cell medium (ESM) {20% FBS, 0.1 mM (1x) NEAA, 1x PSQ), 100 μM 2-ME, 1x human Leukemia Inhibitory Factor (hLIF) in IMDM} were used instead of EFM at the step of final medium change of day 6 (see Fig. 2, Fig. 4 upper diagram). The medium was changed every other day. On day 8, the cells were passaged onto feeder cells (MEF cells treated with 15 μg/mL of mitomycin C for 3 hr) plated at a density of 1×106 cells/10-cm dish one day before usage.
Transduction of MEF cells with oriP/EBNA-1 plasmid vectors was performed essentially following the protocol as previously described (Barry, 2004). The MEF cells were seeded in EFM and cultured overnight. They were stripped by trypsin-EDTA treatment and dispersed in RPMI1640 at 3.6×106 cells/300 μL and placed into sterile 0.4-cm gapped cuvette. Then, 5 μg each of tandem O-S and K-M or 10 μg of polycistronic OKSM) were mixed with 0.2 μg of LT and they were added to the cell suspension. Single pulse electroporation was performed with 290 V, 950 μF, at R.T. by Gene Pulser XcellTM (Bio-Rad Laboratories, Hercules, USA). Then, 700 μL of EFM was added to the cuvette and all the suspension was passaged onto feeder cells in 10-cm dish. The medium was changed every other day. On day 9, the medium was changed to ES with conditioned medium (ES C.M.) {ESM with 1/5 volume of a conditioned medium (prepared from a culture medium of MEF in ESM)}.
Isolation of mouse iPSC colonies essentially followed the protocol distributed by the Center for iPS Cell Research and Application (CiRA), Kyoto University (Takahashi et al., 2007). Transduced MEF cells were cultured on feeder cells, changing the media every other day. When colonies were observed (day 20–35), they were each picked using a sterile micropipette tip and transferred separately to 10 μL of 0.25% Trypsin-EDTA in 96-well plate and incubated for 5 min at 37°C. After ninety microliters of ESM was added to each well and the cells suspended by pipetting, they were transferred to 500 μL of ESM in 48-well plate (which were previously plated by feeder cells at a density of 2.5×104 cells/well one day before usage). The clonal iPS cell lines were maintained by passaging every two days in ESM. Freeze stocks were made using BambankerTM (Lymphotec, Tokyo, Japan) at early stages of maintenance.
The character of the iPS cell lines was initially validated by alkaline phosphatase staining and then by immunofluorescent analyses using Nanog and SSEA-1 as pluripotency markers. For the alkaline phosphatase assay, iPS cells, which were passaged in 48-well plates, were assayed by using Alkaline phosphatase kit (Sigma-Aldrich, St. Louis, USA). For the immunofluorescense assay, Anti-SSEA-1, rabbit (Millipore, Billerica, USA) and Anti-Nanog, mouse (Reprocell Inc., Yokohama, Japan) were used as primary antibodies, and then Anti-rabbit IgG, Alexa Fluor® 568 Conjugate and Anti-mouse IgM, Alexa Fluor® 488 Conjugate (Life Technologies, Carlsbad, USA) were used as secondary antibodies. The conditions of the immunofluorescense analysis followed a protocol described in a previous report (Zhao et al., 2009). For the EOS-reporter assay, we used EOS-S(4+)-EmGFP reporter (Hotta et al., 2009b) manufactured as BacMam viruses (Fig. 1d; kindly provided by Drs. Uma Lakshmipathy and Pauline Lieu of Life Technologies, R&D) in order to mark effectively reprogrammed iPS cells. The results of immunofluorescent analysis and EOS-reporter assay were obtained by fluorescent microscopy by using a fluorescence microscope (Eclipse TE2000-E; Nikon) with filter sets for CFP/YFP/Cy5 (86008v1; Chroma Technology Corp.), BEF/GFP/DsRed (86009; Chroma Technology Corp.).
For the teratoma assay, 0.5×106 cells/20 μl of iPS cells in HBSS was injected into the testis of SCID mice (CLEA Japan, Inc. Tokyo, Japan). Five to six weeks after injection, tumors were processed by Bouin’s fixative and analyzed histologically.
We are grateful to Dr. Uma Lakshmipathy (Life Technologies) for providing valuable information for construction of BacMam system, and Dr. Pauline Lieu (Life Technologies) for providing EOS-S(4+)-EmGFP viral particles and information on BacMam transduction. We also thank Dr. James Ellis for providing EOS(C3+) and EOS(S4+) genes. This work was supported in part by a Grant-in-Aid for Scientific Research from The New Energy and Industrial Technology Development Organization, Japan (NEDO). This work was carried out at the Life Technologies Corporation-Endowed Laboratory and financial support in part by a fostering fund of Life Technologies (Invitrogen) Corp. on the development of Multisite Gateway DNA cloning system. Gateway Platinum, pCR, TOPO, BigDye, One Shot, Max Efficiency, Library Efficiency, Cellfectin and Alexa Fluor are registered trademarks of Life Technologies. Clonase, PureLink, pDONR, DH10B, DH10Bac, Mach1, Premo, pENTR and pDEST are trademarks of Life Technologies.
|