| Kouhei Takashima, Akina Saitoh: These authors contributed equally to this work. To whom correspondence should be addressed: Kazuhisa Nakayama, Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. Tel/Fax: +81–75–753–4527/4557 E-mail: kazunaka@pharm.kyoto-u.ac.jp Abbreviations: Arf, ADP-ribosylation factor; ATGL, adipose triglyceride lipase; BFA, brefeldin A; COPI, coat protein I; COPII, coat protein II; ER, endoplasmic reticulum; ERGIC, ER-Golgi intermediate compartment; FBS, fetal bovine serum; GAP, GTPase-activating protein; GCA, golgicide A; GEF, guanine nucleotide exchange factor; LD, lipid droplet; MDCK, Madin-Darby canine kidney; RNAi, RNA interference; siRNA, small interfering RNA; SREBP, sterol regulatory element binding protein; TGN, trans-Golgi network; TIP47, tail-interacting protein of 47 kDa. |
Various aspects of membrane traffic are regulated by small GTPases. Among them, those belonging to the ADP-ribosylation factor (Arf) family primarily regulate budding of coated carrier vesicles, such as COPI-coated vesicles from the cis face of the Golgi apparatus and clathrin/AP-1-coated vesicles from the trans-Golgi network (TGN) and endosomes (D’Souza-Schorey and Chavrier, 2006; Gillingham and Munro, 2007). There are five Arf isoforms in humans (Arf1-Arf6; humans lack Arf2), which are grouped into three classes on the basis of sequence similarity: class I, Arf1 and Arf3; class II, Arf4 and Arf5; and class III, Arf6. Among them, class I and class II Arfs have been implicated primarily in anterograde and retrograde transport processes from the Golgi (Volpicelli-Daley et al., 2005).
Arfs regulate coated vesicle formation through switching between a GDP-bound inactive state and a GTP-bound active state. GDP-bound Arfs are predominantly cytosolic, while, upon activation, GTP-bound Arfs are recruited onto membranes and in turn recruit coat proteins, such as the COPI coat complex at the cis-Golgi and the AP-1 adaptor complex at the TGN, and induce membrane budding. Exchange of GDP on Arfs for GTP is catalyzed by guanine nucleotide exchange factors (GEFs), which share a Sec7-like catalytic domain. In humans, there are 14 Arf-GEFs (Casanova, 2007; Gillingham and Munro, 2007); among these Arf-GEFs, GBF1 regulates, through activating Arfs, formation of COPI-coated vesicles at the cis-Golgi and endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC), while BIG1 and BIG2 regulate formation of clathrin/AP-1-coated vesicles at the TGN and endosomes (Shin and Nakayama, 2004). On the other hand, hydrolysis of bound GTP to GDP and inorganic phosphate is stimulated by GTPase-activating proteins (GAPs). The human genome encodes 31 Arf-GAPs, which share a zinc finger-like Arf-GAP domain (Gillingham and Munro, 2007; Inoue and Randazzo, 2007; Kahn et al., 2008); among these Arf-GAPs, we have recently shown that ArfGAP1, ArfGAP2 and ArfGAP3 redundantly regulates COPI-mediated retrograde transport from the cis-Golgi to the ER (Saitoh et al., 2009).
Over the course of the above experiments, we noticed that cells triple-depleted of ArfGAP1, ArfGAP2 and ArfGAP3 by small interfering RNAs (siRNAs) share some phenotypes with those of cells depleted of β-COP, a subunit of the COPI coat complex. One of the common phenotypes is exaggerated vacuolar structures (Saitoh et al., 2009). On the other hand, recent studies by systematic RNA interference (RNAi) experiments revealed that knockdown of components involved in COPI-mediated retrograde transport promoted lipid droplet (LD) formation (Beller et al., 2008; Guo et al., 2008), and a recent proteomic study indicated the presence of ArfGAP1 as well as Arfs and COPI subunits on LDs (Bartz et al., 2007). Taken into account these data, we speculated that knockdown of Arfs and their regulators, which are involved in COPI-mediated Golgi-to-ER retrograde transport, also increases lipid storage. In this study, we show by siRNA experiments and using inhibitors of Arf-GEFs that disturbance of components of the GBF1-Arf-COPI-ArfGAP machinery that are involved in the Golgi-to-ER retrograde transport increases lipid storage.
HeLa and MDCK (Madin-Darby canine kidney) cells were cultured in minimum essential medium eagle (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen). HuH-7 cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich) supplemented with 10% heat-inactivated FBS or lipoprotein-deficient FBS (Biomedical Technologies).
Polyclonal rabbit antibodies to GBF1, BIG1, BIG2 and ArfGAP1 were prepared as described previously (Ishizaki et al., 2006; Kawamoto et al., 2002; Saitoh et al., 2009; Shin et al., 2004). Polyclonal rabbit anti-β-COP antibody was purchased from Thermo Scientific. Monoclonal mouse antibodies against γ-adaptin and Sec31A were from Sigma-Aldrich and BD Biosciences, respectively. Polyclonal rabbit anti-Arf4 and monoclonal mouse anti-Arf5 antibodies were from ProteinTech Group and Abnova, respectively. Polyclonal guinea pig anti-TIP47 antibody was from Progen. Monoclonal rat anti-HA antibody was from Roche Applied Science. AlexaFlour- and horseradish peroxidase-conjugated secondary antibodies were from Molecular Probes and Jackson ImmunoResearch Laboratories, respectively. Golgicide A (GCA) was synthesized based on the procedure described by Sáenz et al. (2009). BODIPY 493/503 was purchased from Molecular Probes. Brefeldin A (BFA) and bovine serum albumin were from Sigma-Aldrich. Saponin and gelatin were from Nacalai Tesque.
An expression vector for C-terminally HA-tagged adipose triglyceride lipase (ATGL) was constructed by subcloning of a cDNA fragment covering entire coding sequence of human ATGL into the pcDNA3-HAC vector (Shin et al., 1997). An S47A mutation was introduced into the ATGL cDNA using a Quik Change Lightning Site-Directed Mutagenesis kit (Stratagene).
Triple knockdown of ArfGAP1, ArfGAP2 and ArfGAP3 (Saitoh et al., 2009), double knockdown of BIG1 and BIG2 (Ishizaki et al., 2006), double knockdown of Arf1 and Arf3 (Man et al., 2011), and single knockdown of β-COP (Saitoh et al., 2009) were performed as described previously. For knockdown of GBF1, Arf4 and Arf5, pools of siRNAs directed for nucleotide residues 1115–2086, 731–1230 and 471–942 (when the A residue of the initiation Met codon is assigned as residue 1) of human mRNAs, respectively, were prepared using a BLOCK-iT RNAi TOPO transcription kit and a BLOCK-iT Dicer RNAi kit (Invitrogen). Cells were transfected with the siRNA pool using Lipofectamine 2000 (Invitrogen) for 24 h. The cells were sub-cultured, incubated for further 24 h, and retransfected with the siRNA pool for 24 h. The cells were then sub-cultured, incubated for 48 h, and subjected to the following analyses.
Cells grown on coverslips were cultured for 6 h or 16 h in serum-free medium containing 400 μM oleic acid. The cells were then fixed with 3% paraformaldehyde for 15 min, treated with 50 mM NH4Cl for 20 min and permeabilized with 0.1% Triton X-100 for 5 min. The cells were incubated with 10 μg/ml BODIPY 493/503 for 1 h to probe LDs or blocked with 10% FBS for 30 min and incubated sequentially with primary and secondary antibodies for 1 h each for immunofluorescence analysis using an Axiovert 200 MAT microscope (Carl Zeiss) or an A1-RMP confocal laser-scanning microscope (Nikon). When cells were stained with both BODIPY 493/503 and antibodies, BODIPY 493/503 was included in the secondary antibody solution. For staining with anti-TIP47 antibody, permeabilization was performed with saponin, in place of Triton X-100, for 10 min and blocking was with 0.2% bovine serum albumin and 0.2% gelatin.
To determine cellular triacylglycerol contents, triacylglycerols were extracted from cells as described by Tsuboi et al. with some modifications (Tsuboi et al., 2004). Cells pre-incubated with oleic acid as described above were trypsinized and collected. The numbers of the collected cells were counted, and ~3.2×106 cells were suspended in 500 μl of isopropanol and sonicated to extract intracellular triacylglycerols. Following centrifugation, a portion (300 μl) of the supernatant was subjected to triacylglycerol quantification using a Triglyceride E-test kit (Wako Pure Chemical Industries).
Cells were lysed in lysis buffer (50 mM Tris-HCl pH7.5, 1% Triton X-100, 150 mM NaCl) containing a protease inhibitor cocktail (EDTA-free) (Nacalai Tesque). The lysates were boiled in SDS sample buffer and separated by 5% SDS-PAGE for Arf-GEFs or 12.5% SDS-PAGE for Arfs. The separated proteins were transferred onto an Immobilon-P transfer membrane (Millipore). The membrane was blocked in 5% skim milk and incubated sequentially with primary and horseradish peroxidase-conjugated secondary antibodies. Detection was carried out using a Chemi-Lumi One L kit (Nacalai Tesque).
In our previous study, we showed that cells triple-depleted of ArfGAP1, ArfGAP2 and ArfGAP3 by siRNAs exhibited phenotypes similar to those of cells depleted of β-COP, a subunit of the COPI coat protein complex (Saitoh et al., 2009). For example, in both the triple ArfGAP-depleted and β-COP-depleted cells, proteins cycling between the cis face of the Golgi apparatus and the ER, such as ERGIC-53, were accumulated at the ERGIC and retrograde transport of the KDEL receptor from the cis-Golgi to the ER was blocked (Saitoh et al., 2009). We then set out to investigate whether the triple ArfGAP depletion affects deposition of LDs for the following reasons: (i) knockdown of subunits of the COPI complex was reported to increase lipid storage (Beller et al., 2008; Guo et al., 2008), although the underlying mechanism has been poorly understood; and (ii) because LDs are generated from the ER by a sequence of events that include accumulation of triacylglycerols and cholesteryl esters between the two leaflets of the ER membrane, gradual growth into a globular shape, and final pinching off from the ER (Martin and Parton, 2006; Ohsaki et al., 2009), it was possible that the Arf-COPI–mediated retrograde transport from the cis-Golgi to the ER could somehow participate in LD biogenesis.
As shown in Fig. 1, treatment of HeLa cells with siRNAs for ArfGAP1+2+3 promoted lipid storage when the cells were incubated with oleic acid; LDs visualized with a lipophilic dye (BODIPY 493/503) were significantly larger than those observed in control cells (compare A and B). The LD deposition was comparable to that observed in β-COP-knockdown cells (C and D). Essentially the same results were obtained with HuH-7 cells (Fig. S1, A–C), which are derived from human hepatocellular carcinoma and have been often used in experiments of LD formation, excluding a possibility that the deposition of LDs by knockdown of ArfGAPs or β-COP is a cell line-dependent phenomenon.
 View Details | Fig. 1. Increase in lipid accumulation by depleting HeLa cells of ArfGAPs or β-COP. HeLa cells treated with a pool of control siRNAs (A, siRNAs for LacZ) or a mixture of siRNAs for ArfGAP1, ArfGAP2 and ArfGAP3 (B) for a total of 96 h, or those treated with a pool of siRNAs for LacZ (C) or β-COP (D) for a total of 48 h were cultured for 16 h in serum-free medium containing 400 μM oleic acid and stained with BODIPY 493/503. The images were acquired with the same gain setting. The same set of siRNA-treated cells were also subjected to triacylglycerol (TG) quantification as described in Materials and Methods (E). The data of ArfGAP siRNA and its control (left two bars) are means±SD of three independent experiments. The data of β-COP siRNA and its control (right two bars) are means of two independent experiments. Note that we have failed to collect more data concerning the triacylglycerol content in β-COP-knockdown cells because viability of cells subjected to the β-COP siRNA treatment was extremely low. |
To support the morphological data, we then measured triacylglycerol contents in the control and knockdown cells. As shown in Fig. 1E, the triple ArfGAP-knockdown caused an approximately 2.5-fold increase in the cellular triacylglycerol content, compared with the control cells. These results suggest that the large LDs in the triple ArfGAP-knockdown cells and in the β-COP-knockdown cells are resulted, at least in part, from the increase in the triacylglycerol content.
Because the Arf small GTPases regulate formation of COPI-coated vesicles by cycling between a GDP-bound inactive state and a GTP-bound active state and knockdown of ArfGAP1+2+3 increases deposition of LDs, like that of β-COP, we then examined whether knockdown of Arf-GEFs also affects lipid storage. In the Golgi region, three high-molecular weight Arf-GEFs have been reported to participate in Arf activation. GBF1 associates predominantly with the cis-Golgi and the ERGIC and participates in trafficking involving COPI-coated vesicles (García-Mata et al., 2003; Kawamoto et al., 2002; Shin and Nakayama, 2004; Zhao et al., 2002), whereas BIG1 and BIG2 associate mainly with the TGN and recycling endosomes and participate in trafficking involving clathrin/AP-1-coated vesicles (Ishizaki et al., 2008; Shin et al., 2004; Shin and Nakayama, 2004; Shinotsuka et al., 2002; Zhao et al., 2002).
We therefore examined whether GBF1 depletion alters lipid storage. At first, we confirmed by immunoblotting that GBF1 was almost completely depleted by treating HeLa cells with a pool of siRNAs for GBF1 (Fig. 2A). Under these conditions, the GBF1 knockdown appeared to promote LD deposition (Fig. 2, compare B and C); LD deposition was also increased in GBF1-knockdown HuH-7 cells (Fig. S1, compare A and D). In parallel with the morphological data, the triacylglycerol content was significantly increased in the GBF1-knockdown HeLa cells (Fig. 2F). In contrast, double knockdown of BIG1 and BIG2 did not significantly affect LD deposition (Fig. 2, D and E) or the triacylglycerol content (Fig. 2F), although both Arf-GEF proteins were efficiently depleted (more than 95%) by the siRNA treatment (Fig. 2A) as described previously (Ishizaki et al., 2006, 2008). These results suggest that impairing the Golgi-to-ER traffic involving GBF1 causes lipid accumulation.
 View Details | Fig. 2. Increase in lipid accumulation by depleting HeLa cells of GBF1. HeLa cells treated with a pool of LacZ siRNAs (B) or GBF1 siRNAs (C) for a total of 120 h, or those treated with a pool of LacZ siRNAs (D) or a mixture of BIG1 and BIG2 siRNAs (E) for a total of 72 h were cultured for 16 h in serum-free medium containing 400 μM oleic acid and stained with BODIPY 493/503. The images were acquired with the same gain setting. The same set of siRNA-treated cells were also subjected to immunoblot analysis with antibody against GBF1, BIG1 or BIG2 (A) or triacylglycerol quantification (F) as described in Materials and Methods. In (F), the data are means±SD of three independent experiments. |
To support the Arf-GEF knockdown data, we then exploited Arf-GEF inhibitors, BFA and golgicide A (GCA), and a cell line possessing the GBF1 protein resistant to these inhibitors. BFA has been widely used in membrane trafficking studies over two decades and inhibits all the three large Arf-GEFs (GBF1, BIG1 and BIG2) (Jackson, 2000; Shin and Nakayama, 2004), while GCA is a recently developed inhibitor specific for GBF1 (Sáenz et al., 2009). On the other hand, MDCK cells have the GBF1 protein that has critical amino acid substitutions in the catalytic domain compared with GBF1 of other species including humans, and thereby does not allow binding of BFA or GCA (Sáenz et al., 2009).
Prior to examining whether these inhibitors differentially affect LD formation in HeLa and MDCK cells, we indirectly confirmed cell line-dependent effects of these inhibitors on GBF1 and BIG1+BIG2 by utilizing differential dependence of membrane association of coat proteins on these Arf-GEFs; the COPI complex is recruited onto cis-Golgi membranes under the regulation of GBF1, while the AP-1 clathrin adaptor complex is recruited onto TGN membranes downstream of BIG1 and BIG2 (Shin and Nakayama, 2004). As shown in Fig. S2, BFA treatment inhibited membrane association of the AP-1 (γ-adaptin) in both HeLa (K and L) and MDCK (W and X) cells, whereas the same treatment inhibited membrane association of β-COP in HeLa cells (I and J) but not in MDCK cells (U and V); these results agree well with those of our previous study (Torii et al., 1995). On the other hand, GCA treatment did not affect γ-adaptin association in HeLa (G and H) or MDCK (S and T) cells. In contrast, the GCA treatment, like BFA treatment, inhibited membrane association of β-COP in HeLa cells (E and F) but not in MDCK cells (Q and R). Thus, these observations indirectly support that GBF1 is sensitive to both BFA and GCA in HeLa cells while it is resistant to these drugs in MDCK cells, and that BIG1 and BIG2 are sensitive to BFA and insensitive to GCA in both HeLa and MDCK cells.
We then compared effects of GCA and BFA on lipid storage in HeLa and MDCK cells. As shown in Fig. 3, A–C, both GCA and BFA appeared to increase LD deposition in HeLa cells. Furthermore, the triacylglycerol content was significantly increased in both GCA-treated and BFA-treated HeLa cells compared with the control cells (G). In contrast, neither GCA nor BFA detectably increased LD deposition (D–F) or the triacylglycerol content (G) in MDCK cells, although the basal triacylglycerol content (mock-treated with DMSO) was higher than that in HeLa cells.
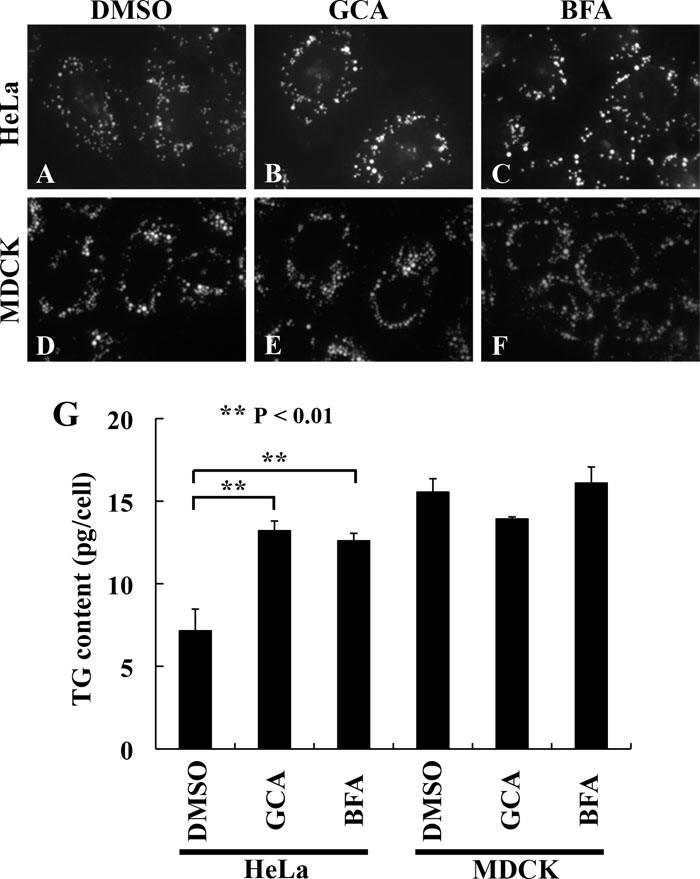 View Details | Fig. 3. Increase in lipid accumulation by chemical inhibition of GBF1 in HeLa cells. HeLa cells (A–C) or MDCK cells (D–F) were mock-treated with DMSO (A and D) or treated with 30 μM GCA (B and E) or 10 μM BFA (C and F) for 6 h in the presence of 400 μM oleic acid and stained with BODIPY 493/503. The images were acquired with the same gain setting. The same set of treated cells were also subjected to triacylglycerol quantification (G) as described in Materials and Methods. The data are means±SD of three independent experiments. |
Together with the siRNA data (Fig. 2), we conclude that depletion/inhibition of GBF1, which is responsible for COPI-mediated retrograde transport from the cis-Golgi to the ER through activating Arfs, results in an increase in LD deposition due, at least in part, to an increase in the triacylglycerol content.
The above-mentioned results together indicate that, under the control of Arfs, COPI-mediated membrane traffic from the cis-Golgi to the ER is implicated in regulation of lipid homeostasis. Which Arf isoform(s) participate in the regulation? We were interested in class II Arfs (Arf4 and Arf5), because a systematic siRNA study of Kahn and colleagues reported that knockdown of Arf4+Arf5 most profoundly affected Golgi-to-ER retrograde transport of the KDEL receptor among knockdowns of Arf combinations examined (Volpicelli-Daley et al., 2005), and because Melançon and colleagues showed that class II Arfs associated with the ERGIC along with GBF1 and suggested that these Arfs regulate membrane trafficking at the ERGIC (Chun et al., 2008). At first, we confirmed specific depletion of Arf4 or Arf5 or both Arf4 and Arf5 by treating cells with pools of siRNAs (Fig. 4A). Under these conditions, single knockdown of Arf4 (Fig. 4C) or Arf5 (Fig. 4D) did not apparently affect LD deposition. However, simultaneous knockdown of Arf4 and Arf5 promoted LD deposition (Fig. 4E) and led to an increase in the cellular triacylglycerol content (Fig. 4H). In contrast, double knockdown of Arf1 and Arf3, which both are class I Arfs that function mainly at the TGN and/or endosomes (Manolea et al., 2010; Volpicelli-Daley et al., 2005), less significantly affected LD deposition (Fig. 4, compare F and G). Together with the fact that the Golgi-to-ER retrograde transport is suppressed in cells knocked down of class II Arfs (Volpicelli-Daley et al., 2005), these results suggest that the Golgi-to-ER retrograde transport under the control of, at least, class II Arfs participates in regulation of lipid homeostasis. We have also attempted to examine effects of triple knockdown of Arf1, Arf4 and Arf5, and simultaneous knockdown of all class I and class II Arfs on lipid homeostasis, because GBF1 is able to activate both class I and class II Arfs (Kawamoto et al., 2002). However, the attempts have so far been unsuccessful because these knockdowns result in an extreme reduction in cell viability (data not shown).
 View Details | Fig. 4. Increase in lipid accumulation by depleting HeLa cells of class II Arfs. HeLa cells treated with a pool of siRNAs for LacZ (B), Arf4 (C), Arf5 (D) or Arf4+Arf5 (E) for a total of 120 h, or a pool of siRNAs for LacZ (F) or Arf1+Arf3 (G) for a total of 96 h were cultured for 16 h in serum-free medium containing 400 μM oleic acid and stained with BODIPY 493/503. The images were acquired with the same gain setting. The same set of siRNA-treated cells in (B)–(E) were also subjected to immunoblot analysis with antibody against Arf4, Arf5 or β-actin (A) or triacylglycerol quantification (H) as described in Materials and Methods. In (H), the data are means±SD of three independent experiments. |
Previous RNAi screening studies (Beller et al., 2008; Guo et al., 2008) and our detailed analyses using siRNAs for components of the GBF1-Arf-COPI-ArfGAP trafficking machinery demonstrate that impairing any component increases deposition of LDs, concomitant with an increase in the cellular triacylglycerol content. On the other hand, a previous proteomic study reported that various proteins implicated in membrane trafficking associated with LDs (Bartz et al., 2007). The proteomic study also reported that Arf-COPI components, including Arf1, Arf4, Arf5, Arf6, α-COP, β-COP and γ-COP, were recruited onto LDs in the presence of a non-hydrolyzable GTP analog, GTPγS, although all of them, but for Arf1, were undetectable in the original fraction of purified droplets (Bartz et al., 2007). We therefore examined whether components of the GBF1-Arf-COPI-ArfGAP machinery associate with LDs by immunofluorescence analysis.
As shown in Fig. 5, however, we failed to find direct association and close apposition of β-COP (A–A''), GBF1 (B–B''), Arf4 (C–C'') and ArfGAP1 (D–D'') with LDs depicted by staining with BODIPY 493/503. We also failed to detect LD association of Sec31A (E–E''), which is a component of the COPII coat complex and a marker of the ER exit sites. Thus, our data show that any of the GBF1-Arf-COPI-ArfGAP components examined is not, at least constitutively, associated with or closely apposed to LDs, and suggest that the GBF1-Arf-COPI-ArfGAP machinery regulates lipid homeostasis by suppressing lipid synthesis and/or stimulating lipolysis indirectly through mediating the retrograde transport, rather than by directly associating with LDs. In view of the prevailing model for LD biogenesis where a portion of membranes containing lipid esters are pinched off from the ER to become isolated LDs (Martin and Parton, 2006; Ohsaki et al., 2009), COPII-mediated anterograde transport and COPI–mediated retrograde transport between the ER and the Golgi may coordinately tune the balance between lipolysis and lipogenesis. One of the possible mechanisms explaining our observations is that the retrograde transport participates in delivery of factors involved in lipolysis to pre-formed LDs or retrieval/sequestration of factors involved in lipid biosynthesis from forming LDs.
 View Details | Fig. 5. None of GBF1-Arf-COPI-ArfGAP components constitutively associates with LDs. HeLa cells cultured for 16 h in serum-free medium containing 400 μM oleic acid were stained with BODIPY 493/503 and processed for immunostaining for β-COP (A-A''), GBF1 (B-B''), Arf4 (C-C''), ArfGAP1 (D-D'') or Sec31A (E-E''). |
While this study was in progress, Soni et al. reported by immunofluorescence analysis that COPI and COPII components decorated the perimeter of LDs (Soni et al., 2009). This was in contrast with our data shown in Fig. 5. Furthermore, they reported that LD association of adipose triglyceride lipase (ATGL), which is a primary triacylglycerol lipase both in adipose and non-adipose tissues (Smirnova et al., 2006; Zechner et al., 2009), was inhibited by treating cells with siRNAs for COPI or COPII components or expression of dominant-negative GBF1 (Soni et al., 2009). On the basis of these results, the authors proposed a model in which ATGL undergoes sequential COPII- and COPI-mediated transport from the ER exit sites through the ERGIC to LDs where it hydrolyzes triacylglycerols (Soni et al., 2009).
We therefore attempted to find the cause of the inconsistency between our data and those of Soni et al. One of the differences between the two studies was that we identified LDs by staining cells with a lipophilic dye (BODIPY 493/503), whereas they detected LDs by immunostaining with antibody to TIP47 (tail-interacting protein of 47 kDa), a protein associated with the LD surface, throughout their study. However, even when we identified LDs by immunostaining with polyclonal guinea pig anti-TIP47 antibody, which was derived from the same source as that used by Soni et al., we failed to find any obvious evidence indicating the constitutive localization of β-COP and GBF1 on the surface or in the vicinity of LDs (Fig. S3).
We then asked whether COPI-mediated transport is responsible for delivery of ATGL onto LDs and in turn for ATGL-mediated lipolysis. Previous studies showed that exogenously expressed ATGL inhibits LD formation in cells incubated with oleic acid (Lu et al., 2010; Smirnova et al., 2006; Yang et al., 2010). We therefore examined whether β-COP depletion competes with exogenously expressed ATGL for diminishing LD formation. As shown in Fig. 6A, a–a''', when control siRNA-treated HeLa cells were transfected with an expression vector for C-terminally HA-tagged wild-type ATGL, the transfected cells (indicated by asterisks) showed markedly diminished LD formation compared with surrounding non-transfected cells; exogenously expressed ATGL(WT)-HA per se exhibited cytoplasmic distribution. Depletion of β-COP by siRNA treatment did not significantly attenuate the inhibitory effect of ATGL(WT)-HA on LD formation (Fig. 6A, b–b'''). These results suggest that exogenously expressed ATGL fulfilled its catalytic function independently of COPI-mediated processes. It remained, however, possible that overexpressed ATGL non-specifically had access to LDs, albeit with a low efficiency, and hydrolyzed triacylglycerols. In order to separate delivery of ATGL onto the LD surface from its catalytic action, we then exploited an S47A mutant of ATGL, which is catalytically inactive but retains an ability to associate with LDs (Lu et al., 2010; Smirnova et al., 2006). When expressed in control cells, ATGL(S47A)-HA associated with LDs (Fig. 6A, c–c'''). In cells depleted of β-COP, the ATGL mutant still distinctly decorated the perimeter of LDs, the size of which was significantly larger than that in control cells (Fig. 6A, d–d'''). Thus, it is likely that COPI-mediated transport makes little, if any, contribution to delivery of ATGL onto the LD surface.
 View Details | Fig. 6. Association of ATGL with LDs is independent of COPI and GBF1. HeLa cells treated with control siRNAs or siRNAs for β-COP for 24 h (A) or GBF1 for 120 h (B) as described in the legends for Fig. 1, C and D, and Fig. 2, B and C, were transfected with an expression vector for C-terminally HA-tagged wild-type ATGL (a-a''' and b-b''') or its S47A mutant (c-c''' and d-d'''). The cells were stained with BODIPY 493/503 (a-d) and processed for double immunostaining with anti-HA antibody (a'-d') and either anti-β-COP or anti-GBF1 antibody (a''-d''). Cells expressing the HA-tagged ATGL construct are marked by asterisks. |
While this manuscript was in preparation, the same research group as Soni et al. further reported that GBF1 interacted with ATGL and suggested that GBF1 could contribute to ATGL delivery onto LDs, although there was no direct evidence linking the GBF1-ATGL interaction to the localization of ATGL on the LD surface (Ellong et al., 2011). To address this issue, we examined whether depletion of GBF1 by siRNA treatment affects lipolysis mediated by exogenously expressed ATGL(WT) and association of ATGL(S47A) with LDs. As shown in Fig. 6B, GBF1 depletion did not significantly relieve the inhibitory effect of exogenously expressed ATGL(WT)-HA on LD formation (compare a–a''' and b–b''') or suppress localization of ATGL(S47A)-HA on the surface of LDs (compare c–c''' and d–d'''). These observations make it unlikely that GBF1 participates directly in association of ATGL with LDs and in ATGL-mediated lipolysis.
Our detailed analyses using siRNAs for components of COPI-mediated transport machinery, in combination with inhibitors of Arf-GEFs, demonstrate that disturbing these components increases deposition of LDs concomitant with an increase in the cellular triacylglycerol content. These components include GBF1 (an Arf-GEF), class II Arfs (Arf4 and Arf5), β-COP (a COPI subunit), and ArfGAP1-ArfGAP3, all of which are involved in retrograde transport from the cis-Golgi to the ER (Chun et al., 2008; García-Mata et al., 2003; Saitoh et al., 2009; Volpicelli-Daley et al., 2005). A prevalent model for LD biogenesis is that triacylglycerols and cholesteryl esters accumulate between the two leaflets of the ER membrane, gradually grow into a globular shape, and are finally pinched off from the ER to become isolated LDs (Martin and Parton, 2006; Ohsaki et al., 2009). In addition, at least in yeasts, LDs have a functional connection with the ER, the connection which allows partitioning of membrane proteins between the two compartments (Jacquier et al., 2011). A previous proteomic study suggested the presence of Arfs and COPI subunits on LDs (Bartz et al., 2007). However, we have failed to show that components of the GBF1-Arf-COPI-ArfGAP transport machinery contact, at least constitutively, with LDs. In addition, although recent studies suggested that the Arf-COPI–mediated transport was involved in delivery of ATGL onto LDs (Soni et al., 2009) and GBF1 might participate in the delivery through directly interacting with ATGL (Ellong et al., 2011), we have found that depletion of β-COP or GBF1 does not significantly affect association of ATGL with LDs or ATGL-mediated lipolysis.
Two general explanations are possible for promotion of lipid storage; namely, down-regulation of lipolysis and up-regulation of lipogenesis. The data presented in this study make it unlikely that enhanced lipid deposition observed in cells depleted of GBF1 or COPI results from suppression of lipolysis, at least that catalyzed by ATGL, although it remains possible that alleviation of lipolysis mediated by other lipolytic enzymes is implicated in the increase in lipid storage. ATGL does not have a transmembrane domain, but instead has a hydrophobic stretch (Zechner et al., 2009), which is required for association of ATGL with LDs and subsequent degradation of LDs (Lu et al., 2010). Therefore, it has been a prevailing view that ATGL is recruited onto LDs directly from the cytosol (Zechner et al., 2009). On the other hand, Soni et al. proposed a model in which sequential actions of COPII-mediated and COPI-mediated membrane trafficking somehow contribute to delivery of ATGL onto LDs (Soni et al., 2009); they presented data indicating that disturbing either COPII or COPI components resulted in delocalization of ATGL from the LD surface. Since COPII-coated vesicles are responsible for transport from the ER to the ERGIC or the cis-Golgi, this model is attractive in view of the fact that LD biogenesis takes place in the ER. However, in contrast to COPI knockdown, knockdown of COPII components has been reported to have no apparent effect on lipid storage (Beller et al., 2008; Soni et al., 2009). Further studies will be necessary to reconcile the apparent discrepancy between the ATGL delivery model and observed effects of COPI and COPII impairment on lipid storage.
On the other hand, what mechanisms are expected to underlie potential up-regulation of lipogenesis? Extracellularly applied fatty acid (oleic acid) is activated to fatty acyl-CoA (oleoyl-CoA) by acyl-CoA synthetase, which associates with the cytoplasmic aspect of the mitochondrial outer membrane, and the acyl-CoA is in turn utilized to esterify diacylglycerols to yield triacylglycerols by diacylglycerol acyltransferases, which are embedded in the ER membrane (Yen et al., 2008). Generally, since resident ER proteins are not always resident in the ER, but rather often escape out of the ER towards the Golgi, these proteins must be retrieved from the Golgi back to the ER by COPI-mediated transport (Lippincott-Schwartz et al., 2000; Lowe and Kreis, 1998). However, if diacylglycerol acyltransferases, which once left the ER, were retrieved by COPI-mediated retrograde transport, impairing the GBF1-Arf-COPI-ArfGAP retrograde transport machinery should result in suppression of lipogenesis.
We give another possible explanation for enhanced lipogenesis under conditions where the COPI-mediated Golgi-to-ER transport is inhibited. This explanation involves SREBP (sterol regulatory element binding protein), a master regulator of lipid biosynthesis. A number of enzymes responsible for biosynthesis of fatty acids and triacylglycerols as well as those involved in cholesterol biosynthesis are under the regulation of SREBP (Horton et al., 2002; Rawson, 2003). Although SREBP is a transcription factor, it resides in the ER as a transmembrane precursor under sterol-rich conditions. Upon a decrease in the cellular sterol level, however, the precursor exits the ER to the Golgi via COPII-coated vesicles and, in the Golgi, undergoes sequential limited proteolysis by site-1 and site-2 proteases to liberate the cytoplasmic domain, which is then translocated into the nucleus and promotes transcription of a number of genes for lipogenic enzymes (Goldstein et al., 2006; Horton et al., 2002; Rawson, 2003). A previous study showed that, when cells were treated with BFA, the SREBP precursor underwent proteolytic processing to yield the mature transcription factor domain irrespectively of the presence of sterols (DeBose-Boyd et al., 1999). Because BFA treatment is known to disintegrate the Golgi apparatus to fuse with the ER (Presley et al., 1998), the site-1 and site-2 proteases are allowed to access the SREBP precursor in the fused ER-Golgi compartment (DeBose-Boyd et al., 1999). It is therefore tempting to speculate that, in the continued presence of BFA, cells can constantly produce mature SREBP, which in turn induces expression of enzymes responsible for biosynthesis of triacylglycerols and cholesteryl esters, leading to deposition of LDs. In contrast to BFA treatment, however, depletion of Arf-COPI components does not give rise to fusion of the Golgi with the ER, although the depletion blocks the Golgi-to-ER retrograde transport (Saitoh et al., 2009), implying that the site-1 or site-2 protease does not have access to the SREBP precursor unless the precursor is transported to the Golgi. Furthermore, in a previous RNAi screening study, knockdown of an SREBP ortholog in Drosophila cells was reported to increase, but not decrease, the size of LDs, although there was no description about the cellular triacylglycerol content (Guo et al., 2008).
Thus, there are advantages and disadvantages with respect to the potential mechanisms underlying enhanced lipid storage observed in cells with impaired GBF1-Arf-COPI-ArfGAP machinery. However, these mechanisms are not mutually exclusive but rather can be coordinated. Further studies to define which proteins involved in lipid metabolism are transported by the GBF1-Arf-COPI-ArfGAP machinery will be a great help towards a better understanding of the mechanism underlying lipid homeostasis regulated by membrane trafficking.
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Japan Society for Promotion of Science, and the Targeted Proteins Research Program.
|