| Edited by Toshihiko Shiroishi. Masayuki Sumida: Corresponding author. E-mail: msumida@hiroshima-u.ac.jp |
Mitochondrial (mt) DNAs of vertebrates are closed circular molecules spanning a length of approximately 16-18 kbp and typically containing 37 genes for 2 ribosomal (r)RNAs, 22 transfer (t)RNAs, and 13 proteins together with the control region (CR) (Boore, 1999). The control region is a long noncoding region that includes signals required for regulating and initiating mtDNA replication and transcription (Wolstenholme, 1992). All 37 genes are arranged in the same relative order in almost all vertebrate species from teleost fishes to eutherian mammals (Anderson et al., 1981; Roe et al., 1985; Tzeng et al., 1992; Boore, 1999).
We know, however, that deviations from the typical gene order are not very rare in vertebrates. In fact, more than 430 complete mtDNA sequences have been determined for vertebrate species (National Center for Biotechnology Information Web-site: http://www.ncbi.nlm.nih. gov/genomes/ORGANELLES/7742.html) and gene rearrangements of the mt genome have been reported for marsupials (Pääbo et al., 1991), birds (Desjardins and Morais, 1990; Quinn and Wilson, 1993; Mindell et al., 1998; Haring et al., 2001), reptiles (Kumazawa and Nishida, 1995; Quinn and Mindell, 1996; Macey et al., 1997; Kumazawa et al., 1998; Kumazawa and Endo, 2004), lampreys (Lee and Kocher, 1995), and teleost fishes (Miya and Nishida, 1999; Inoue et al., 2001; Miya et al., 2001; Inoue et al., 2003). Most of these cases involve only a few rearrangements of tRNA genes and/or the control region and occur in a lineage-specific manner.
The complete mtDNA sequences have been reported for 38 amphibian species, including 7 caecilians (Zardoya and Meyer, 2000; San Mauro et al., 2004a), 23 salamanders (Zardoya et al., 2003; Zhang et al., 2003a, b; Mueller et al., 2004), and the following 6 anurans: three discoglossid frogs, Alytes obstetricans, Bombina orientalis, and Discoglossus galganoi (San Mauro et al., 2004b), the clawed toad Xenopus laevis (family Pipidae) (Roe et al., 1985), the Japanese pond frog Rana nigromaculata (family Ranidae) (Sumida et al., 2001) and Buergeria buergeri (family Rhacophoridae) (Sano et al., 2004). In anurans, X. laevis and three discoglossid frogs share the conserved vertebrate gene order containing only the tRNA-Phe gene between the control region and the 12S rRNA gene. However, the R. nigromaculata mt genome contains four tRNA genes (tRNA-Leu(CUN), tRNA-Thr, tRNA-Pro, and tRNA-Phe) in this region. This tRNA gene cluster is also present in B. buergeri. In addition, the ND5 gene is located bet-ween the control region and the tRNA gene cluster in B. buergeri.
To further our understanding of the evolution of mt genome structures in Rhacophoridae, we determined the complete mtDNA nucleotide sequence of Schlegel’s tree frog, Rhacophorus schlegelii. The genus Rhacophorus is regarded as a member of the subfamily Rhacophorinae, whereas the genus Buergeria is classified as a member of the subfamily Buergerinae, which is thought to be the most basal group in Rhacophoridae (Wilkinson and Drewes, 2000). We describe our observations on the characteristics of the Rh. schlegelii mt genome, including the novel finding that it has two control regions and a unique gene rearrangement. The evolutionary implications of our findings are also discussed.
Schlegel’s tree frog (Rhacophorus schlegelii) was collected in the suburbs of Hiroshima City, Hiroshima Prefecture, Western Japan. The total genomic DNA was extracted from a clipped toe of this specimen using a DNeasy Tissue Kit (QUIAGEN).
The DNA sequence determination strategy for the Rh. schlegelii mt genome was the same as that used for Buergeria buergeri (Sano et al., 2004). A partial mt segment from the Cytb gene to the ND5 gene was initially amplified by long-and-accurate PCR (LA-PCR) and nested PCR methods, using Rh. schlegelii total DNA as the template. PCR mixtures were prepared with a TaKaRa LA Taq™ Kit (TaKaRa). The first PCR reaction was carried out with the primer set CytbFow1-1 and ND5Rev2 (Table 1 and Fig. 1). Using the first PCR product as a template, the second PCR was carried out with the primer set CytbFow1 and ND5Rev1 (Table 1 and Fig. 1). These primers were designed as degenerate primers based on conserved sequences of known amphibian mtDNAs. Both the first and second PCR reactions consisted of an initial denaturation at 94°C for 1 min, 33 cycles of denaturation at 94°C for 20 sec plus annealing and extension at 60°C for 5 min, and a final extension at 72°C for 10 min. The resultant PCR fragment was purified by polyethylene glycol precipitation and used for sequencing. The sequencing was performed using 373A and 3100-Avant automated DNA sequencers (ABI) with DYEnamic ET Terminator cycle sequencing reagent (Amersham) and BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Using the sequence information, the primer ND5Fow_sch/leu was synthesized from the 3’end region of this product (Table 1 and Fig. 1). LA-PCR with ND5Fow_sch/leu and R17N1 gave a product of approximately 4.5 kbp (Table 1 and Fig. 1). During the sequencing of the above two amplified fragments, we found that these fragments were difficult to sequence by primer walking due to an abundance of lengthy tandem repeat units. To determine the precise sequence of these regions, PCR products were cloned using a TOPO TA Cloning Kit (Invitrogen) and a series of deleted subclones were made from each clone using the Exonuclease III deletion method (Henikoff, 1987). The remaining mtDNA fragment was amplified with the primer set R.sch_12S.80_Fow and R.sch_cytb. 400_Rev (Table 1 and Fig. 1) designed based on the 12SrRNA gene and Cytb gene sequences determined above. The amplified mtDNA, a fragment of about 12.5 kbp, was electrophoresed on a 0.7% agarose gel, and thereafter the DNAs were purified from the excised gel using a GenElute Agarose Spin Column (SIGMA) and both strands were directly sequenced by the primer walking method. The complete nucleotide sequence of the Rh. schlegelii mtDNA was deposited in the DDBJ database under accession number AB202078.
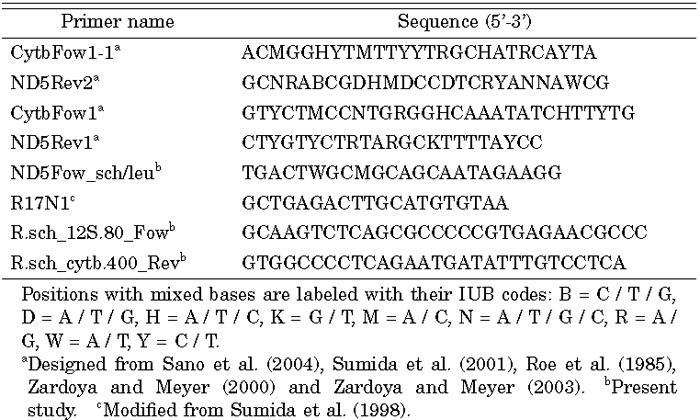 View Details | Table 1. Primers used for PCR amplification in the present study |
 View Details | Fig. 1. Sequencing strategy for Rhacophorus schlegelii mtDNA. Localization and directions of primers used in the LA-PCR amplification and DNA sequencing are denoted by arrowheads. LA-PCR products are shown as bold lines below the gene map. |
For the phylogenetic analysis, we created an alignment dataset using CLUSTAL W (Thompson et al., 1994). The data set consisted of all 37 mt gene sequences of 4 anurans, 1 salamander (Mertensiella luschani), and 1 caecilian (Typhlonectes natans). The latter two were used as outgroups. The alignment was revised by eye, and all positions with gaps and ambiguous sites were excluded. Based on the alignment data (13,437 nucleotide sites), we reconstructed a phylogenic tree by the maximum likelihood (ML) method. The tree construction was performed with PAUP* ver. 4.10b (Swofford, 2001). The best-fit model of DNA substitution was estimated using ModelTest ver. 3.06 (Posada and Crandall, 1998) and a general-time-reversible + gamma + invariant (GTR + G + I, G = 0.9557, I = 0.2978) model was proposed under AIC consideration.
According to our determination of the complete nucleotide sequence of Rh. schlegelii mtDNA, the total length of the genome was 21,359 bp. This was longer than the reported mt genomes of other anurans, which range from 17,014 bp (Discoglossus galganoi) to 19,959 bp (Buergeria buergeri). Though the mt genome of Rh. schlegelii was extremely large, it contained only the 37 genes typically found in other vertebrates; i. e., 13 protein genes, 2 rRNA genes, and 22 tRNA genes (Fig. 2 and Table 2), all of which were similar in length to their counterpart genes in other anurans. The considerable difference in genome size is due to a duplication of the control region and the accumulation of lengthy repetitive sequences in the control regions (see the section on the Noncoding region).
 View Details | Fig. 2. Organization of the Rhacophorus schlegelii mitochondrial genome. All protein-coding genes are encoded by the H strand, with the exception of ND6, which is encoded by the L strand. Transfer RNA genes are represented by the standard one-letter amino acid code, and those encoded by the H and L strands are shown outside and inside the circle, respectively. L1, L2, S1, and S2 represent tRNA-Leu(CUN), tRNA-Leu(UUR), tRNA-Ser(UCN), and tRNA-Ser(AGY), respectively. Asterisks indicate lengthy intergenic spacers. Genes are abbreviated as in Table 2, except for A6 and A8, which indicate ATPase6 and ATPase8, respectively. CR1 and CR2 indicate control regions 1 and 2, respectively. |
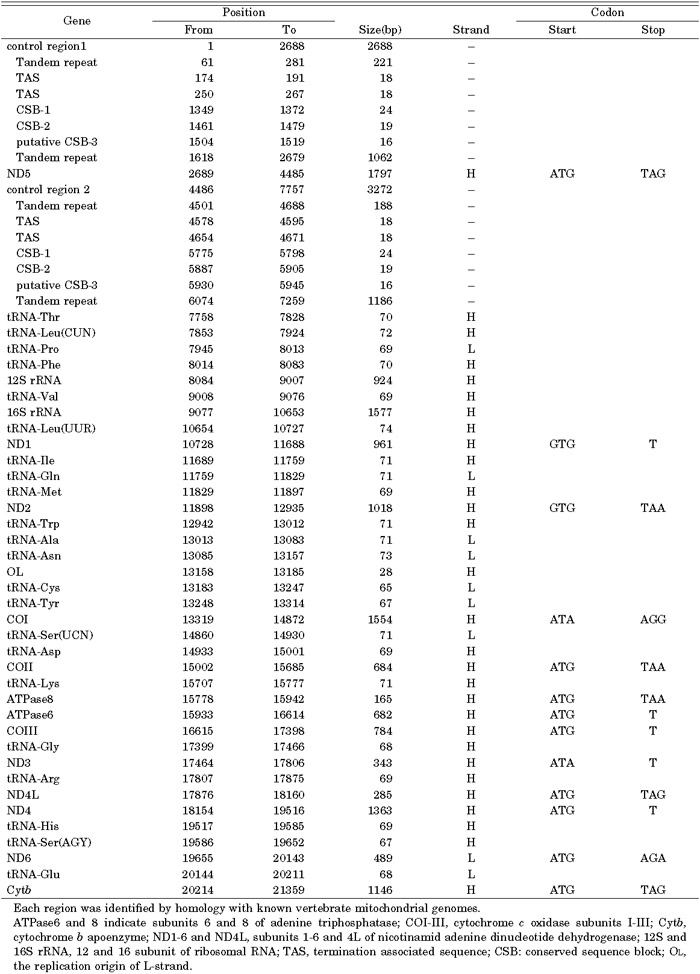 View Details | Table 2. Location of features in the mitochondrial genome of Rhacophorus schlegelii |
The base composition of the complete Rh. schlegelii mtDNA was A: 31.8%, T: 30.3%, G: 14.7%, and C: 23.3%. The slightly high A + T content (62.1%) of Rh. schlegelii is similar to those of other vertebrates.
The mt gene arrangement of Rh. schlegelii diverged from those of typical vertebrates. The ND5 gene and four tRNA genes (tRNA-Thr, tRNA-Leu(CUN), tRNA-Pro and tRNA-Phe) were located between the control region and 12S rRNA gene in the Rh. schlegelii mt genome (Fig. 3). The ND5 gene and four tRNA genes were also located in this region of the B. buergeri mt genome. In Rh. schlegelii, on the other hand, the positions of the tRNA-Thr and tRNA-Leu(CUN) genes were exchanged compared to those of B. buergeri and an additional control region was found between the ND5 gene and tRNA-Thr gene. The presence of two control regions has not been observed in the mt genomes of any of the other anurans so far reported.
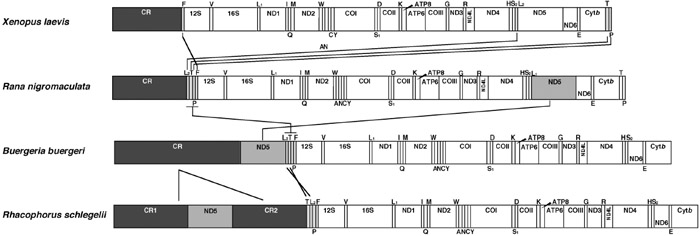 View Details | Fig. 3. Comparison of mitochondrial gene arrangements among Rhacophorus schlegelii, Buergeria buergeri, Rana nigromaculata (Sumida et al., 2001) and Xenopus laevis (Roe et al., 1985). Arrows indicate the rearranged homologous genes. Closed and shaded boxes indicate the genes whose positions vary. Genes are abbreviated as in Table 2 and Fig. 2. |
All of the 13 protein-coding genes found in other animals were also present in the Rh. schlegelii mt genome. The codon usage of the genome was identical to that of other vertebrates. All but two of these genes started with an ATG or ATA initiation codon (the two exceptions, ND1 and ND2, both started with a GTG codon). Two genes (COI and ND6) ended with the AGR (AGG and AGA) stop codon characteristic of vertebrate mt genomes. Six protein genes (ATPase8, COII, Cytb, ND2, ND5 and ND4L) stopped with the usual TAR codon. Each of the remaining 5 genes (ATPase6, COIII, ND1, ND3 and ND4) had an incomplete stop codon, a single stop nucleotide T, where the post transcriptional polyadenylation could produce a complete TAA stop codon (Ojala et al., 1980).
The 12S and 16S rRNA genes in Rh. schlegelii were 924 and 1,577 bp in length, respectively. As in other vertebrates, these rRNA genes were situated between the tRNA-Phe and tRNA-Leu(UUR) genes and separated by the tRNA-Val gene.
Twenty-two tRNA genes were identified in the mt genome of Rh. schlegelii by comparison with homologues of other amphibians. Twenty-one of the 22 tRNAs could be folded into the canonical cloverleaf secondary structure (Fig. 4). The one exception, tRNA-Ser(AGY), had an unpaired dihydrouridine (D) arm. The unpaired D-arm in tRNA-Ser(AGY) is a common feature of metazoan mt genomes (Wolstenholme, 1992).
 View Details | Fig. 4. Secondary structure representation of Rh. schlegelii mitochondrial tRNA genes. Watson-Crick base pairing is indicated by solid bars (–), and the G-T pairs that commonly occur in animal mt genomes are shown with plus marks (+). |
Two major noncoding regions were found in the Rh. schlegelii mt genome. One was a 2,688 bp region located between the Cytb and ND5 genes in a position homologous to that of the control region in the B. buergeri mt genome. The other was a 3,237 bp region located between the ND5 and tRNA-Thr genes (Fig. 2). These regions were thought to correspond to the control regions, as each contained several components characteristic of the control regions such as a termination-associated sequence (TAS) and two conserved sequence blocks (CSB-1 and CSB-2) (Fig. 5). A similar candidate sequence for CSB-3 (putative CSB-3) was also found in the control regions (Fig. 5). We termed these control regions CR1 (between the Cytb and ND5 genes) and CR2 (between the ND5 and tRNA-Thr genes) (Fig. 2). Both CR1 and CR2 possessed tandem repeats at both the 5’ and 3’ sides. The 5’ repeated regions consisted of 5 complete repeat units of 40 bp in CR1 and 4 complete repeat units of 40 bp in CR2. The repeated motif at the 5’ side showed high sequence similarity in the two control regions, whereas the repeated motif at the 3’ side and the number of 3’ side repeats differed between the two control regions. The 3’ repeated regions consisted of 97 repeat units of 11 bp in CR1 and 27 repeat units of 18 bp and 35 repeat units of 20 bp in CR2. Furthermore, nonrepetitive sequences of approximately 500 bp were found in CR2 between the 3’-side tandem repeat regions and tRNA-Thr gene. CR1 and CR2 could be aligned in a span of about 1,510 bp from the 101st nucleotide (Fig. 5) and showed high nucleotide similarity (97%). This aligned section encompassed TAS and all of the conserved sequence blocks.
 View Details | Fig. 5. Alignment of the duplicated control regions of the Rh. schlegelii mt genome. The sequences are presented with alignment between comparable regions of the two control regions (CR1 and CR2). Repeat units are shown by underlines. Conserved sequence elements are boxed. Abbreviations: TAS, termination associated sequence; CSB, conserved sequence block. Dots indicate nucleotides identical between CR1 and CR2. Dashes indicate gap sites introduced in the sequence to optimize the alignment. |
The origin of L-strand replication (OL) of the Rh. schlegelii mt genome was located between the tRNA-Asn and tRNA-Cys genes in the WANCY tRNA gene cluster (Fig. 2). The OL was 28 nucleotides in length and the sequence had the potential to fold in a stem-loop secondary structure with a stem formed by 11 pairs of nucleotides and a loop of 6 nucleotides. The “GCCGG” conserved motif characteristic of vertebrate OL was also found at the base of the stem within the tRNA-Cys gene in Rh. schlegelii.
In the Rh. schlegelii mt genome, two short noncoding sequences of 24 and 20 bp were found adjacent to both the 5’ and 3’ ends of the tRNA-Leu(CUN) gene (Fig. 2). The 24-bp noncoding sequence was located upstream of the tRNA-Leu(CUN) gene and had 70.8% similarity to the tRNA-Pro gene sequence. The 20-bp noncoding sequ-ence had 70.0% similarity to the tRNA-Thr gene sequ-ence.
We analyzed the phylogenetic relationship among anurans using the long nucleotide sequences of all the mt genes. The resultant ML tree (-lnL = 63417.20573) is shown in Fig. 6. The robustness of our result was confirmed by high bootstrap support of all nodes. Xenopus laevis (family Pipidae) was the first to branch away, Rana nigromaculata (family Ranidae) was the second, and thereafter the rhacophorid frogs, B. buergeri and Rh. schlegelii, formed a monophyletic group (=family Rhacophoridae).
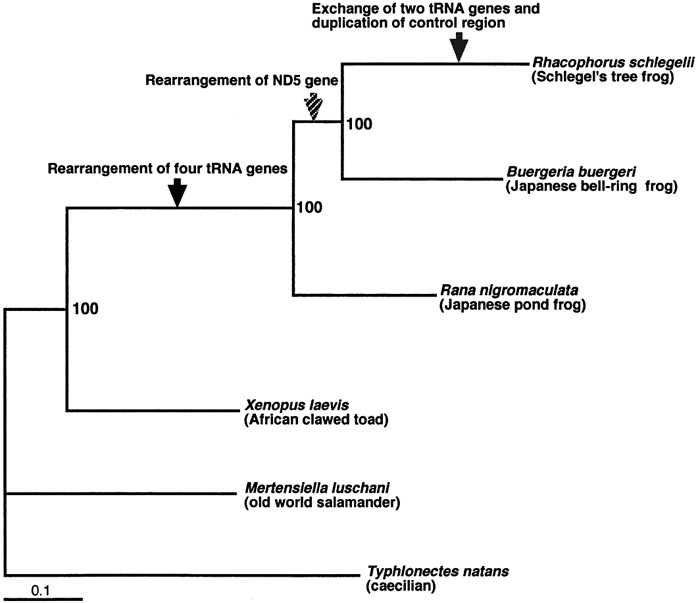 View Details | Fig. 6. Maximum-likelihood (ML) tree based on all 37 mitochondrial genes (13,437 nucleotide sites). The following parameters were used for the ML analysis: the proportion of invariable sites (I) = 0.2978, gamma distribution shape parameter (α) = 0.9557, empirical base frequencies (A: 0.3071; C: 0.2613; G: 0.1582; T: 0.2734), and substitution rates ([A–C] = 2.6087, [A–G] = 5.4442, [A–T] = 2.6111, [C–G] = 0.5089, [C–T] = 9.5809, [G–T] = 1). The scale bar shows the genetic distance calculated using the above parameter settings. The values of internal branches are the bootstrap values (1,000 replicates). The following accession numbers were used for the sequences: Buergeria buergeri (AB127977; Sano et al., 2004), Rana nigromaculata (AB043889; Sumida et al., 2001), Xenopus laevis (M10217; Roe et al., 1985), Mertensiella luschani (AF154053; Zardoya and Meyer, 2003) and Typhlonectes natans (AF154051; Zardoya and Meyer, 2000). The latter two were used as outgroups. Arrows indicate the inferred lineages in which gene rearrangement events occurred (see text). |
The Rh. schlegelii mt genome possessed two control regions with 97% sequence similarity over 1,510 bp (Fig. 5). Our newest findings suggest that the rhacophorid Polypedates leucomystax also shares duplicated control regions (Sano et al., in preparation). The P. leucomystax mt genome includes duplicated control regions with an identical arrangement to that in Rh. schlegelii. While the duplicated control region sequences have great similarity to each other, they differ considerably between these two species (<70%). This evidence suggests that the duplicated control regions are undergoing concerted evolution. The existence of two control regions has been reported in the following metazoans: snakes (Kumazawa et al., 1998), Komodo dragon (Kumazawa and Endo, 2004), parrots (Eberhard et al., 2001), fishes (Lee et al., 2001, Inoue et al., 2003), sea cucumbers (Arndt and Smith, 1998), and metastriate ticks (Black and Roehrdanz, 1998). High sequence similarity between duplicated control regions has been observed in all of these cases. If there are two control regions with similar sequences, this could be a hot target for slipped-strand mispairing (Levinson et al., 1987), the process responsible for the tandem duplication of the spacing region between the two control regions. Kumazawa et al. (1998) suggested that the replacement of a control region resulting from such a tandem duplication event may lead to homogeneity of the control regions (Fig. 7).
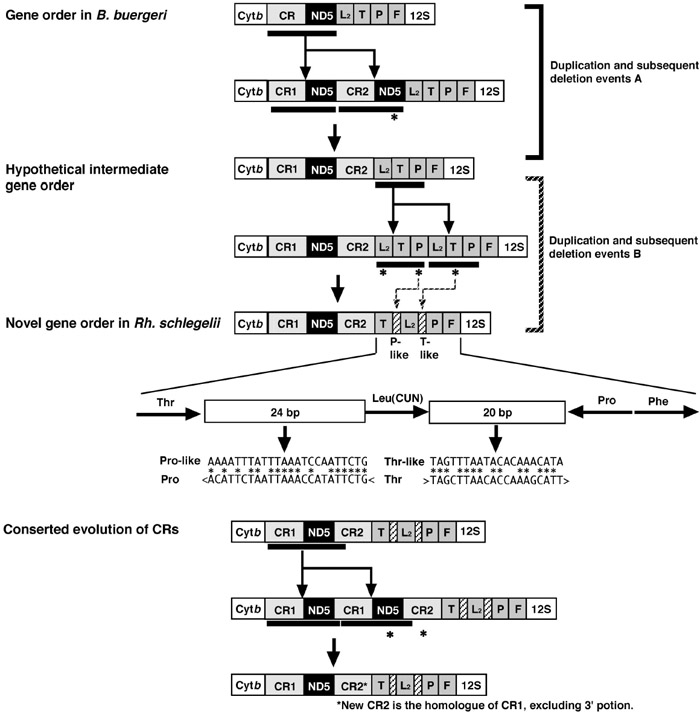 View Details | Fig. 7. Possible gene rearrangement pathways from the gene order of B. buergeri to that of Rh. schlegelii through a hypothetical intermediate gene order, and the concerted sequence evolution of the control regions. Thick bars below genes denote a duplication unit whose endpoints are uncertain. Deleted genes and control region are shown with asterisks. Genes are abbreviated as in Table 2 and Fig. 2. Sequence alignments are shown between the tRNA-Pro-like sequence and the partial tRNA-Pro gene sequences (positions 1-24), and between the tRNA-Thr-like sequence and the partial tRNA-Thr gene sequences (positions 8-27). Right- or left-angle brackets indicate the transcriptional direction of each gene. |
The present study showed that the gene order of the Rh. schlegelii mt genome differs from that typical in vertebrates but shares similarity to that of B. buergeri, another species from the same family, Rhacophoridae. Only two differences were observed between the gene arrangements of Rh. schlegelii and B. buergeri: exchange of the tRNA-Leu(CUN) and tRNA-Thr genes and an additional control region in Rh. schlegelii. The genus Buergeria is regarded as the most basal group in the family Rhacophoridae (Channing, 1989; Jiang et al., 1987; Liem, 1970; Richards and Moore, 1998; Wilkinson and Drewes, 2000; Wilkinson et al., 2002). Furthermore, the gene order of B. buergeri can be derived from that of R. nigromaculata by a single event (Sano et al., 2004), whereas that of Rh. schlegelii needs at least 3 rearrangement events. Our newest findings also reveal that the gene order of B. buergeri is shared by other rhacophorid frog, Kurixalus eiffingeri (Sano et al., in preparation). We can therefore reasonably infer that the gene arrangement of the B. buergeri mt genome is an ancestral condition of rhacophorid frogs, and by extension we can speculate that the gene arrangement of Rh. schlegelii is derived from the ancestral condition. Gene rearrangement in the animal mt genome is generally believed to take place through tandem duplication of gene regions as a result of slipped strand mispairing followed by multiple deletions of redundant genes (Moritz and Brown, 1986, 1987; Moritz et al., 1987; Boore and Brown, 1998). According to the gene rearrangement mechanism, at least two duplication-deletion events must have occurred to explain the rearrangement that changed the gene order of B. buergeri to that of the Rh. schlegelii (Fig. 7). One is a tandem duplication in the region which contains the control region and the ND5 gene followed by deletion of the ND5 gene (Fig. 7A). The other is a tandem duplication occurring in the region which contains the tRNA-Leu(CUN), tRNA-Thr, and tRNA-Pro genes followed by deletions of redundant genes (Fig. 7B). The presence of two short noncoding sequences adjacent to both the 5’ and 3’ ends of the tRNA-Leu(CUN) gene seems to prove this rearrangement pathway. These noncoding sequences showed some similarity to the tRNA genes associated with gene rearrangement (Fig. 7). However, it remains unclear which event (A or B in Fig. 7) occurred more recently in the evolution of the Rhacophoridae mt genomes. Further investigation of rhacophorid frogs may provide data to elucidate the evolution of rhacophorid mt gene arrangement in more detail.
In the present report we showed that the cluster of four tRNA genes previously known in R. nigromaculata and B. buergeri is also shared by Rh. schlegelii. But in the case of Rh. schlegelii, two of the tRNA genes are arranged in different orders. Rhacophorus schlegelii also has two control regions separated by the ND5 gene. We were also interested to find that another rhacophorid group (Polypedates) shares the gene arrangement of Rh. schlegelii (Sano et al., in preparation). Family Rhacophoridae has been categorized into two subfamilies, Buergerinae and Rhacophorinae (Duellman, 1993). Although Buergerinae is a monotypic category that accommodates the relatively small genus Buergeria, the intergeneric relationships of Rhacophorine genera (e.g., Chirixalus, Philautus, Polypedates, Rhacophorus, and Theloderma) remain unresolved (Liem, 1970; Channing, 1989; Dubois, 1986; Wilkinson et al., 2002). The mt genome organization found here in Rh. schlegelii may be one of useful characteristics for elucidating the phylogenetic relationships of rhacophorid frogs.
This work was supported by Grants-in-Aid for Scientific Research to M. Sumida (No. 13839012) and A. Kurabayashi (No. 16770066) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and a Grant-in-Aid for the Extensive Research Program of Hiroshima Prefectural Government to T. Fujii.
|