| Edited by Minoru Murata. Satoshi Kitamura: Corresponding author. E-mail: kitamura@taka.jaeri.go.jp |
Polyploidization by genome doubling is an almost ubiquitous event in plants (Wendel, 2000). This event is one of the factors that broaden genetic diversity through the following mutations and genomic rearrangements, and is therefore important in a speciation mechanism (Soltis, 2005). Nicotiana species comprise two categories with respect to their cytogenetic features: one consists of species with 2n = 24 and their descendants with 2n = 18~20, and the other consists of species with 2n = 48 and their descendants with 2n = 32~46 (Goodspeed, 1954). It is likely that species in the former category are diploid, whereas those in the latter category are derived from amphidiploidization of the 2n = 24 species. However, genomic lineages in the 2n = 48 group are mostly unclear: the speciation of the 2n = 48 group appears to be more complex due to putative recurrent hybridization or introgression events among ancestral species, as well as possible chromosome fusion (Goodspeed, 1954). The 2n = 48 species and their descendants possess some useful and desired characters such as disease- and insect-resistance (Stavely, 1979; Bai et al., 1995). Elucidation of the genome composition and genomic relationships is important for understanding the evolutionary processes of Nicotiana species and for more effectively using their useful characters in breeding programs.
Nicotiana tabacum has long been used as a model plant for genetics, cell biology, and molecular biology. The N. tabacum genome is thought to be amphidiploid based on morphological and cytological observations, biochemical characteristics, composition of gene families and of repeated sequences (Goodspeed, 1954; Gray et al., 1974; Bland et al., 1985; Okamuro and Goldberg, 1985; Matassi et al., 1991; Skalická et al., 2003, 2005). Genomic in situ hybridization (GISH) has provided reliable evidence for the amphidiploidy of N. tabacum, in which the genome is comprised of two subgenomes, S from N. sylvestris and T from N. tomentosiformis (Kenton et al., 1993b; Kitamura et al., 2000; Lim et al., 2000). Because GISH uses total genomic DNA, this molecular cytogenetical technique is a powerful tool for identifying the genome composition in plants (Kenton et al., 1993a). However, GISH analyses using arbitrary probe/chromosome combinations are quite laborious and time consuming. Therefore, molecular indicators that properly reflect genome relationships have been desired. Two kinds of rDNA (45S and 5S rDNA) are prerequisite for plant life, and are allocated as distinctive tandem arrays in the plant genome. Chromosomal sites of the rDNA are frequently used to compare genomes of taxonomically related species (Fukui et al., 1998; Kitamura et al., 2000). Furthermore, it is known that coding regions of the rDNA sequences are conserved while the intergenic spacer regions are moderately divergent among closely related species (Fedoroff, 1979). These characteristics allowed us to use moderately divergent sequences of the rDNA spacer regions for analyzing the interspecific relationships in many plant species (Scoles et al., 1988; Kellogg and Appels, 1995; Liston et al., 1999).
Prior to the recent advance in molecular genetics, Goodspeed (1954) deduced interspecific relationships among Nicotiana species based on the comprehensive investigations for morphological and cytological characteristics. Several recent studies have examined the molecular phylogenetics among Nicotiana species including putative amphidiploid/tetraploid species. These phylogenies have been based on plastid genes (Aoki and Ito, 2000; Clarkson et al., 2004) and internal transcribed spacer (ITS) sequences of 45S rDNA (Chase et al., 2003). However, relationships at the genome level are quite limited (Chase et al., 2003). We previously described the interspecific relationships among 2n = 24 species and their descendants in Nicotiana and detected genomic lineages between well-known amphidiploid species and their parental species, based on the chromosomal localization and sequence information of the intergenic spacer region of the 5S rDNA (Kitamura et al., 2001). Here, we applied this analytical program to genome-unidentified 2n = 48 species and their descendants to obtain insights into their genomic origin. Analysis on the 5S rDNA provided some clues for inferring the relationships between 2n = 24 and 2n = 48 species. These relationships were also examined by GISH at the genome level.
Wild tobacco species including Nicotiana repanda, N. quadrivalvis, N. umbratica, N. debneyi, N. gossei, N. suaveolens, and N. africana were used in this experiment. The chromosome numbers and current taxonomic classifications of these species are shown in Table 1. The nomenclature and the current taxonomic classifications are according to the recent revision by Knapp et al. (2004). For genome analysis, interspecific hybrids between N. paniculata and N. langsdorffii were obtained by artificial cross pollination. These plants were cultivated in a greenhouse maintained at about 25°C under natural light conditions.
 View Details | Table 1. Number of chromosomes and rDNA sites in Nicotiana species used in this study. The comparable diploid species (see text) are also included |
Total genomic DNA was extracted from fresh, mature leaves using the CTAB procedure (Murray and Thompson, 1980). Using genomic DNA as template, PCR amplification of the intergenic spacer sequence of the 5S rDNA was performed with a primer pair named 5SrDNA-3 and 5SrDNA-4. Sequences of the primers and the exact PCR protocol were described previously (Kitamura et al., 2001).
On the mitotic chromosome spreads, FISH was carried out using 5S and 45S rDNA probes, essentially according to Kitamura et al. (2000). For GISH, genomic DNA labeled with either biotin-16-dUTP or digoxigenin-11-dUTP was used as a hybridization probe. Hybridization, washing, and detection procedures were the same as those of Kitamura et al. (2000).
The major PCR band of each species was recovered from an agarose gel and cloned into pGEM-T easy vector (Promega, Madison, USA). At least five clones were completely sequenced for each fragment. Because only a slight sequence heterogeneity was found among the randomly-selected five clones, consensus sequences were developed for respective PCR bands by the majority rule at each nucleotide site.
Multiple alignment was carried out using CLUSTAL W (Thompson et al., 1994). A neighbor-joining tree was constructed using the PHYLIP package (Felsenstein, 1995). A maximum parsimony tree was also constructed using heuristic search settings in PAUP ver. 3.1 (Swofford, 1993). Internal support of the clades was assessed using the bootstrap procedure of 100 replicates.
The coding region of the 5S rDNA was used as a probe for the FISH analysis, and the hybridization sites were detected as green signals in Fig. 1. For N. repanda and N. quadrivalvis, one metacentric chromosome pair showed a strong hybridization signal at the (sub)terminal region among the 48 chromosomes (Fig. 1a and 1b). In addition to one minor locus, one major 5S rDNA locus was detected near the centromere of an acrocentric pair of N. debneyi (Fig. 1d). On one chromosome pair out of 23 pairs of N. africana, a clear hybridization signal was detected at an intercalary position (Fig. 1g). Two major 5S rDNA loci were found for N. gossei (Fig. 1e) and N. suaveolens (Fig. 1f), although the morphology of the chromosomes harboring 5S rDNA was apparently different between these two species, and one minor locus was often found in N. suaveolens. For N. umbratica, one major 5S rDNA locus at an intercalary region of a submetacentric chromosome and three minor loci at the subterminal regions of acrocentric chromosomes were detected (Fig. 1c). The numbers of the 5S rDNA and 45S rDNA sites determined in our analysis are given in Table 1. It seems likely that minor rDNA sites have DNA sequences with relatively low homology to the probe DNA and/or with low copy number of arrays compared to the major rDNA sites. We could not yet conclude whether minor rDNA sites reflect degradation of the previously-present major rDNA sites or generation of new sites.
 View Details | Fig. 1. Chromosomal localization of 5S rDNA (green signals) on mitotic chromosomes. (a) N. repanda; (b) N. quadrivalvis; (c) N. umbratica; (d) N. debneyi; (e) N. gossei; (f) N. suaveolens; (g) N. africana. Major and minor 5S rDNA loci are indicated by arrowheads and open arrowheads, respectively. Scale bars indicate 10 μm. |
Using a primer set specific for the amplification of the 5S rDNA spacer sequence (Kitamura et al., 2001), PCR was carried out for seven wild-tobacco species. Although some minor fragments were amplified, one major band with the strongest intensity was found for each species. The fragment sizes were 500 bp for N. repanda, 510 bp for N. quadrivalvis, 180 bp for N. umbratica, 520 bp for N. debneyi, 180 bp for N. gossei, 300 bp for N. suaveolens, and 600 bp for N. africana. The DNA fragment recovered from the selected PCR band was cloned into a TA-cloning vector. Sequencing of at least five clones per PCR band did not reveal any large insertions/deletions, and showed low sequence variability within species. Accordingly, the consensus sequence for each species was easily constructed.
Multiple alignment with the respective consensus sequences as well as those for some 2n = 24 species (Kitamura et al., 2001) allowed us to identify some large deletions (Fig. 2), assuming that the consensus sequence of the 5S rDNA spacer in Nicotiana was about 600 bp in length (Kitamura et al., 2001; Matyásek et al., 2002). N. suaveolens, N. repanda, N. quadrivalvis, and N. debneyi had species-specific large deletions, while N. gossei and N. umbratica shared one common large deletion (about 400 bp in length) (Fig. 2). In downstream end of the deletion in N. gossei and N. umbratica, a common sequence (AGTTT) was found.
 View Details | Fig. 2. Schematic representation of large deletions (> 20 bp) from the consensus sequence of the 5S rDNA in Nicotiana. Primers for the amplification of the spacer sequence were indicated by facing arrowheads, which are located directly below their positions in the diagram above. Three regions (I~III) and one subregion (III-A) of the spacer sequence, which were previously defined in diploid Nicotiana species (Kitamura et al., 2001), are also indicated. |
Because the 3'-region of the spacer (about 300 bp in length) is relatively conserved and without large deletions, for all of the species except N. suaveolens, N. gossei and N. umbratica there is enough to compare the patterns of nucleotide exchanges (Fig. 2). This region corresponds to Region III, which was previously defined as a relatively conserved region among the 5S rDNA spacer sequences from 2n = 24 species and their descendants (Kitamura et al., 2001). Region III was used to evaluate the relationships among respective consensus sequences constructed here, as well as those from 2n = 24 species (Kitamura et al., 2001) (Fig. 3). By neighbor-joining analysis, four putative tetraploid/amphidiploid species distinctively clustered with the diploid species (Fig. 4). N. quadrivalvis and N. repanda grouped together with N. obtusifolia (2n = 24) (referred to as N. trigonophylla in Kitamura et al., 2001), a North American species of the Trygonophyllae section. N. africana grouped with N. langsdorffii (2n = 18), a species of the Alatae section. N. debneyi grouped with N. glauca (2n = 24), a species of the Noctiflorae section. In addition, N. tabacum (600 bp), N. tomentosiformis, N. kawakamii (600 bp), and N. otophora (614 bp) were observed as monophyletic group in the same phylogenetic tree (Fig. 4). This close relationship among the four species is consistent with the findings of previous reports (Kitamura et al., 2000, 2001; Lim et al., 2000; Nakamura et al., 2001; Matyásek et al., 2002). Similar clustering patterns were observed by maximum parsimony analysis (data not shown).
 View Details | Fig. 3. Aligned nucleotide sequences of the 5'-flanking sequences of the gene region of 5S rDNA in Nicotiana. Dashes indicate alignment gaps. Asterisks indicate positions of invariable nucleotides. +1 indicates transcription start point. Because two types of sequences have been isolated from some species (Kitamura et al., 2001), a representative sequence for each species is included in the analysis and its sequence length is also given. AGTTT sequence in N. gossei and N. umbratica is indicated by underline. |
 View Details | Fig. 4. Neighbor-joining tree based on the 5'-flanking sequences of the gene region of 5S rDNA in Nicotiana. Some consensus sequences from each of the other Nicotiana species (Kitamura et al., 2001; Kitamura et al., unpublished data) are also included in the analysis, and are marked with asterisks. Bootstrap values (more than 50%) are indicated at each fork. |
To assess whether the relationships between 2n = 24 and 2n = 48 groups suggested by 5S rDNA reflect the genomic lineages between species, hybridization patterns were investigated using genomic DNA as probes on mitotic chromosomes (Fig. 5).
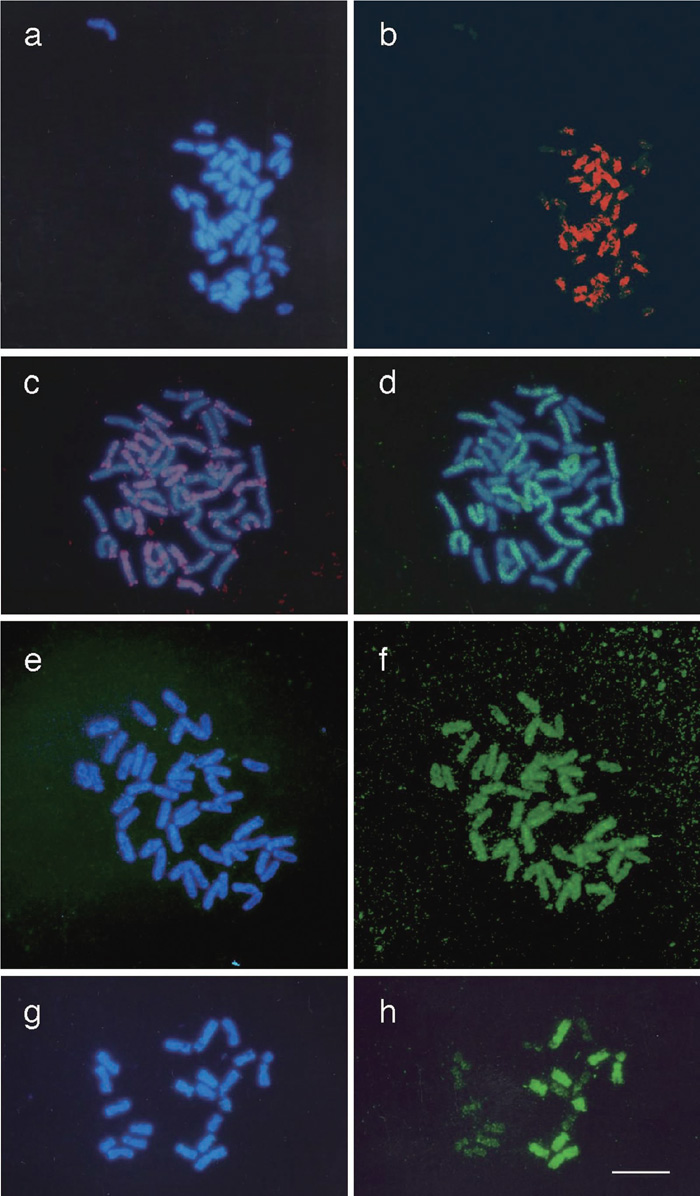 View Details | Fig. 5. Genome analysis of N. debneyi, N. quadrivalvis, and N. africana. (a–b) Mitotic chromosomes of N. debneyi (DAPI image in a) were hybridized with N. glauca genomic DNA (b). (c–d) Mitotic chromosomes of N. quadrivalvis were hybridized with N. obtusifolia genomic DNA (c). After de-staining, N. sylvestris genomic DNA was sequentially hybridized to the same chromosomal spread (d). (e–f) Mitotic chromosomes of N. africana (DAPI image in e) were hybridized with N. langsdorffii genomic DNA (f). (g–h) Mitotic chromosomes of interspecific hybrid between N. paniculata and N. langsdorffii (DAPI image in g) were hybridized with N. africana genomic DNA (h). Scale bar indicates 10 μm. |
Although the hybridization signal was mottled, about half of the 48 chromosomes of N. debneyi were preferentially hybridized with total genomic DNA from N. glauca (Fig. 5a and 5b). For N. quadrivalvis, hybridization signals with N. obtusifolia DNA were detected along with the entire lengths of nearly half of the chromosome set, as well as some regional signals (Fig. 5c). Because N. quadrivalvis was considered to be related to Alatae species based on traditional observations (Goodspeed, 1954), hybridization patterns were determined using genomic DNA of N. sylvestris, as a representative of the Alatae element (Reed, 1991), as another probe for the same chromosomal spread. N. sylvestris DNA was clearly hybridized to the remaining 24 chromosomes out of 48 chromosomes of N. quadrivalvis (Fig. 5d).
Total genomic DNA from N. langsdorffii was strongly hybridized to all of the chromosomes of N. africana (Fig. 5e and 5f). To determine the specificity of the genomic homology, chromosomal spreads of hybrids between N. paniculata and N. langsdorffii were also used as target materials for labeled N. africana DNA. N. paniculata has 24 chromosomes, whereas N. langsdorffii has 18 chromosomes. So the interspecific hybrid between them has 21 chromosomes (Fig. 5g). GISH analysis demonstrated that N. africana genomic DNA was clearly hybridized to 9 chromosomes out of 21 (Fig. 5h). These 9 chromosomes appeared to be derived from N. langsdorffii based on the karyotype of N. langsdorffii (Goodspeed, 1954; Kitamura et al., 2001).
Chromosomal localizations of rDNAs are helpful for inferring genome identity (Bisht and Mukai, 2000). However, data only by FISH are insufficient to evaluate the inter-genomic relationships in the putative amphidiploid Nicotiana species. We previously used the 5S rDNA spacer sequences combined with their chromosomal localizations, to show the interspecific relationships among diploid Nicotiana species (Kitamura et al., 2001). In addition, we also detected genomic lineages in two well-known amphidiploid species, N. tabacum and N. rustica, using 5S rDNA as an indicator (Kitamura et al., 2001). Then, to obtain insights into the genomic origin of other putative amphidiploid species, we applied a similar procedure to the putative amphidiploid species. As a result, we obtained some clues to reveal interspecific relationships between 2n = 24 and 2n = 48 species, and also detected these interspecific relationships by GISH at the genome level. The genomic lineages proposed in this study are discussed below in terms of other molecular phylogenetic results from the literature.
We showed a close relationship between N. debneyi and N. glauca with a high bootstrap value (Fig. 4). Traditional observations led to speculation that N. debneyi originated from an amphidiploid between two parental groups, an early Alatoid (present Alatae or Sylvestres sections) and an early Acuminatoid (present Petunioides section, Table 1, 2) (Goodspeed, 1954; Reed, 1991). One of the two subgenomes in N. debneyi was recently discriminated by GISH with genomic DNA of N. longiflora or N. plumbaginifolia (both are Alatae species) but not with genomic DNA of Petunioides species (Chase et al., 2003) (Table 2). In the present study, N. glauca DNA was preferentially hybridized to a subset of the N. debneyi genome, although this preference between subgenomes was low (Fig. 5a and 5b). This result suggests that one subset of the N. debneyi genome is highly homologous but the other subset is somewhat homologous to the N. glauca genome. N. glauca belonged to the Paniculatae section under the previous classification (Reed, 1991). However, Knapp et al. (2004) recently proposed that N. glauca is a member of Noctiflorae, because molecular phylogenetics using other DNA sequences such as the ITS region of 45S rDNA (nuclear DNA) and the chloroplast DNA sequences also showed that N. glauca was more closely related to Noctiflorae than to Paniculatae and Petunioides (Aoki and Ito, 2000; Chase et al., 2003; Clarkson et al., 2004). These results support the hypothesis that N. glauca or another Noctiflorae species is the most probable candidate for one parent of N. debneyi, in combination with an Alatae ancestor as the other parent.
 View Details | Table 2. Putative origins of Nicotiana amphidiploid species suggested by Goodspeed (1954) and Chase et al. (2003) |
Most of the species in Nicotiana are endemic to South America and Australia, and North American wild-tobacco species are in a minority. Among 2n = 48 species and their descendants, the Polydicliae section (two species) and the Repandae section (four species) are composed only of North American species. Goodspeed (1954) predicted that N. quadrivalvis, a species of Polydicliae, was derived from an amphidiploid between a progenitor of N. attenuata and an early Alatoid species (Table 2). Our GISH pattern (Fig. 5d) implies that N. sylvestris is a candidate for this Alatoid element, but Chase et al. (2003) pointed out that genomic DNA of N. sylvestris and that of N. attenuata were hybridized to the same subgenome of N. quadrivalvis. In the present study, the relationship of N. obtusifolia with N. quadrivalvis was suggested by (i) 5S rDNA spacer sequences (Fig. 4), (ii) similar chromosomal sites of the 5S rDNA (Fig. 1b) (Kitamura et al., 2001), and (iii) the GISH analysis (Fig. 5c). These results indicate that a species closely related to N. obtusifolia is involved in speciation of N. quadrivalvis. Phylogenetic trees based on the plastid genome have been suggested to reflect the maternal lineage in Nicotiana, such as the maternal lineage between N. tabacum and N. sylvestris (Olmstead and Palmer, 1991; Aoki and Ito, 2000; Clarkson et al., 2004). Because a close relationship between N. obtusifolia and N. quadrivalvis was also suggested by the DNA sequences of the maternally inherited plastid genome (Aoki and Ito, 2000; Clarkson et al., 2004), N. obtusifolia appears to be the maternal parent of N. quadrivalvis, and a species with a genome similar to that of N. sylvestris appears to be the paternal parent.
The origin of N. africana has not been fully examined. Based on the ITS sequences of 45S rDNA, N. africana grouped with other Australian species of the Suaveolentes section, and this group formed a cluster with Alatae species with an 82% bootstrap value (Chase et al., 2003). Such phylogenetic relationships have also recently suggested by using plastid DNA sequences (Clarkson et al., 2004). Our results based on 5S rDNA spacer sequences showed that N. africana was a sister species of N. langsdorffii, an Alatae species with 2n = 18 (Table 1, Fig. 4). The chromosomal sites of the 5S rDNA (intercalary location of the long arm) were similar to each other (Kitamura et al., 2001). The close relationship between these species was demonstrated to occur throughout the genome (Fig. 5e–h). As in the case of the Australian species, genomic DNA of two Alatae species with 2n = 20, N. longiflora or N. plumbaginifolia, was found to be hybridized to half of the chromosome set in N. africana, although the difference in hybridization signal between hybridized- and non-hybridized-chromosomes was not described (Chase et al., 2003). These results raise the possibility that N. africana is derived from an amphidiploid, and that both of its parents are closely related to the present-day N. langsdorffii. As N. africana is the only species known to be in Africa (Merxmüller and Buttler, 1975) and its phylogenetic relationships with other species have been examined by only a few study (Chase et al., 2003; Clarkson et al., 2004), more data are needed to clarify its evolution and speciation.
Based on the 5S rDNA spacer sequences, the species used in our study were divided into some groups consisting of the 2n = 24 and 2n = 48 level species (Fig. 4). Chromosomal localizations of the 5S rDNA arrays were similar in most groups. GISH analysis showed clear genomic lineages in each group (Fig. 5). Our results may be related to the characteristics of the 5S rDNA. The 5S rDNA spacer sequences were found to hardly change by gene conversion in the amphidiploid genome (Cronn et al., 1996; Fulnecek et al., 2002; Matyásek et al., 2002), in contrast to frequent gene conversion of the ITS region of 45S rDNA (Wendel et al., 1995; Volkov et al., 1999). Our results indicate that the 5S rDNA spacer sequence is one of the best molecular indicators to reveal genomic lineages in polyploid genera, such as Nicotiana, Gossypium (Cronn et al., 1996), and Triticeae (Baum and Bailey, 2001). Isolation of the 5S rDNA from many other Nicotiana species would help to understand the phylogenetic relationships more accurately in this complex genus.
We wish to thank Masanori Hatashita (The Wakasa Wan Energy Research Center) for helping with the sequence analysis. We also thank Japan Tobacco Inc. for supplying wild-tobacco seeds. We are grateful to James Raymond for careful review of the manuscript.
|