| Edited by Masa-Toshi Yamamoto. Yuji Kageyama: Corresponding author. E-mail: kageyama@bs.naist.jp |
RNA interference (RNAi) was originally applied in Caenorhabditis elegans (Fire et al., 1998) and has since been developed as a powerful tool to elucidate gene function in diverse organisms, including mammals, insects and plants (Hannon, 2002). In each case, RNAi is achieved by using double-stranded RNA (dsRNA) as a ‘trigger’ molecule that recognizes a complementary ‘target’ sequence and eventually induces gene silencing mediated by the RNAi inducing silencing complex (Sontheimer, 2005; Tomari and Zamore, 2005). In mammals, only short dsRNAs (~29 bp) can act as trigger molecules (Elbashir et al., 2001), since longer dsRNAs activate the interferon pathway and result in non-specific silencing of gene expression (Stark et al., 1998), whereas in many other organisms long dsRNAs (~1000 bp) can also induce RNAi.
dsRNA has been introduced by direct application to cells or by establishing transgenic individuals. In Drosophila melanogaster, RNAi can be achieved by two strategies: exogenous RNAi and endogenous RNAi (Carthew, 2003). The exogenous method employs introduction of siRNA into embryos or cultured cells (Hammond et al., 2000; Kennerdell and Carthew, 1998), while the endogenous method involves establishment of transgenic lines expressing a long inverted repeat sequence, which leads to formation of hairpin-type dsRNAs in vivo (Fortier and Belote, 2000; Kennerdell and Carthew, 2000). Although the former method is more rapid and convenient, endogenous RNAi exhibits more stable expressivity and penetrance and is more suitable for many studies, especially for those of postembryonic development. However, construction of plasmids including a long inverted repeat of a target sequence is often problematic; for example, it is widely known that plasmids with a long inverted repeat frequently undergo internal deletion during bacterial culture. To solve this problem, a short linker sequence is commonly placed between the repeated sequences. In several Drosophila RNAi vectors, a functional intron used as the linker sequence increases the effectiveness of RNAi (Lee and Carthew, 2003; Reichhart et al., 2002), presumably because the exon-exon junction complex can promote translocation of the dsRNA into the cytoplasm, the major site of the RNAi pathway. Another difficulty arises from the fact that cloning two identical sequences in opposite orientations limits the use of the multiple cloning sites of a recipient vector plasmid. Recent studies of plant RNAi resolved this problem by using an in vitro recombination system to construct an inverted repeat and placing the target DNA fragment in opposite orientations (Miki and Shimamoto, 2004; Wesley et al., 2001).
Here, we report a novel transformation vector, pRISE, for RNAi experiments in Drosophila. pRISE contains a characteristic repeat of the Gateway recombination cassette, enabling us to construct RNAi transgenes much more easily. pRISE effectively silenced target genes both in Drosophila S2 cells and in transgenic flies, indicating that it is a powerful and convenient tool to elucidate gene function in Drosophila.
The reading frame cassette A (RfA: the attR1-cmr-ccdB-attR2 fragment in the Invitrogen Gateway Vector Conversion System) was cloned into the progenitor plasmid pUAST (Brand and Perrimon, 1993) that had been digested by XhoI and filled by T4 DNA polymerase. The resulting plasmid (referred to as pUAST-RfA) was used in the following pRISE construction.
For pRISE-ftz, a functional ftz intron was amplified from a ftz genomic clone using primers ftz-XbaI (5’-TTTTTCTAGAAGGTAGGCATCACACACGATTAAC-3’) and ftz-rev-SalI (5’-CACAGTCGACCTGTAAGCATAAGCAAAGAAAAAATGGG-3’). The attR1-ccdB-attR2 fragment was also amplified from the RfA using primers RfA-XbaI (5’-GCATCTAGAACAAGTTTGTACAAAAAAGCTGAACG-3’) and RfA-rev-ftz (5’-ATGCTTACAGGTCGACTGTGACCACTTTGTACAAGAAAGCTGAAC-3’). These two fragments were gel-purified and used as templates in a PCR reaction to prepare an RfA-ftz fragment, using the ftz-XbaI and RfA-XbaI primers. The amplified 1.9-kb fragment was gel-purified and cloned into pCR-BluntII-TOPO (Invitrogen). The insert was sequenced, excised by XbaI digestion and ligated to pUAST-RfA. The resulting plasmid was partially sequenced to confirm the orientation of the RfA-ftz fragment and designated pRISE-ftz.
The CS-2 intron was amplified from Drosophila genomic DNA using specific primers (5’-GGATCCAGGTAAGTGGGAGTCGCTTCCTGCAGAATTG-3’/5’-AGATCTCCTGAAAAAAAAAACAGATGCAGGTGGGTTTAGTTTAAGCTCCG-3’), cloned into pCR-BluntII-TOPO and sequenced. pUAST-R57 (GenBank no. AB233207), which contains the Ret intron, was kindly supplied by Dr. Ryu Ueda (National Institute of Genetics, Mishima). For pRISE-Ret and pRISE-CS2, the same strategy was used except that intron-specific primers were used for plasmid construction: Ret-XbaI (5’-TGGTCTAGAGAGTTAAAGGTGGGTAAGAG-3’)/Ret-rev (5’-GATTCCGGAGCCATCCACTTTA-3’) and CS2-XbaI (5’-TGGTCTAGAAGGTAAGTGGGAGTCGCTTCCTGCAGAATTG-3’)/CS2-rev-BglII (5’-AGATCTCCTGAAAAAAAAAACAGATGCAGGTGGGTTTAGTTTAAGCTCCG-3’) primer pairs were used for the Ret and CS-2 introns, respectively. The Ret-XbaI and CS2-XbaI primers were also used to amplify RfA-Ret and RfA-CS2 fragments, respectively, in combination with the RfA-XbaI primer.
pWAGAL4 (Ishikawa et al., 1999; Usui et al., 1999), a GAL4 expression plasmid, was kindly provided by Dr. Yasushi Hiromi (National Institute of Genetics, Mishima).
To construct the plasmids pUAST-EGFP and pUAST-DsRed, the EGFP and DsRed-Express ORFs were amplified from pEGFP-N1 (Clontech) and pDsRed-Express (Clontech) by PCR using specific primer pairs (EGFP: 5’-CACCATGGTCGACCATGGTGAGCAAGGGCGAGGA-3’/5’-TTAGTCGACGCCGCCCTTGTACAGCTCGTCCATGCCGAG-3’; DsRed-Express: 5’-CACCCGCCACCATGGCCTCCTCCG-3’/5’-CTACAGGAACAGGTGGTGGC-3’). The amplified fragments were cloned into pENTR/D-TOPO (Invitrogen) according to the supplier’s instructions. Following confirmation by sequencing, each ORF was cloned into pUAST-RfA (see above) by in vitro recombination using LR Clonase (Invitrogen).
To construct pRISE-ds-EGFP plasmids, a 500-bp fragment of the EGFP ORF was amplified from pEGFP-N1 with specific primers (5’-CACCATGGTGAGCTAGGGCGAGGA-3’/5’-TTGAAGGTTCACCTTGATGCC-3’) and cloned into pENTR/D-TOPO. Following confirmation by sequencing, the trigger fragment of EGFP was integrated into pRISE-ftz, -Ret, or -CS2 by in vitro recombination. The resulting plasmids were designated pRISE-ftz-ds-EGFP, pRISE-Ret-ds-EGFP and pRISE-CS2-ds-EGFP, respectively. pRISE-ds-white was constructed by the same strategy used for pRISE-ds-EGFP, except that specific primers (5’-GCTGAATGCCCTTGCCTTT-3’/5’-CACCTGCCCAAGAAAGCTACCCT-3’) were used to amplify a 554-bp fragment of the white gene from pUAST.
Drosophila S2 cells, kindly provided by Dr. Mikiko Shiomi (Tokushima University), were cultured in Schneider’s Drosophila medium (GIBCO) with 10% fetal bovine serum, 1% penicillin and 1% streptomycin. For RNAi assay, 2 × 105 cells in 500 μl culture medium were transfected with 0.2 μg of pRISE-ds-EGFP or empty pUAST in Cellfectin Reagent (Invitrogen), as well as 0.2 μg each of pWAGAL4, pUAST-EGFP and pUAST-DsRed. Two days after transfection, EGFP fluorescence was measured using an Axiovert fluorescent microscope (Carl Zeiss) and AxioCam (Carl Zeiss). Colors in figures were modified by Photoshop CS and Photoshop Elements 2.0 (Adobe).
Three days after transfection, RNA was prepared from S2 cells using Isogen (Nippon Gene) according to the supplier’s instructions. Five μg total RNA was applied to each lane for electrophoresis, after which RNA was transferred to a BioDYNE nylon filter (Pall) and hybridized with a digoxigenin (DIG)-labeled DNA probe using standard procedures.
DIG-labeled DNA probes for Northern blot analyses were prepared by nested PCR reactions. Template DNA for EGFP or rp49 was first amplified with specific primers, and the amplified fragment was then labeled by secondary PCR using the same primers and DIG-dUTP (Roche). For EGFP, a 220-bp DNA fragment that does not overlap with the RNAi target sequence was amplified from pEGFP-N1 using specific primers (5’-GATCCGCCACAACATCGAGG-3’/5’-TTAGTCGACGCCGCCCTTGTACAGCTCGTCCATGCCGAG-3’). For rp49, the full-length ORF of the rp49 gene was amplified by RT-PCR using specific primers (5’-TCCTTCCAGCTTCAAGATGAC-3’/5’-GTGTATTCCGACCACGTTACA-3’) and cloned into pCRII-TOPO (Invitrogen). The resulting plasmid, pCR-rp49-3, was used as a template for nested PCR with specific primers (5’-TCCTACCAGCTTCAAGATGAC-3’/5’-GTGTATTCCGACCACGTTACA-3’).
Flies were reared at room temperature on standard glucose/cornmeal/yeast media. All mutations are described in Flybase (http://flybase.net/) unless otherwise mentioned. UAS-ds-white transgenic lines were produced by P-element-mediated transformation (Spradling and Rubin, 1982).
To accomplish rapid construction of dsRNA transgenes for RNAi experiments in Drosophila, we developed a novel transformation vector, pRISE (RNAi inducing silencing effector; Fig. 1). This vector contains a characteristic inverted repeat of the attR1-cmr-ccdB-attR2 recombination cassette, enabling us to insert the same target sequence in both orientations using Gateway Technology (Invitrogen). If a trigger sequence is cloned into an appropriate entry vector, such as pENTR/D-TOPO (Invitrogen), it is placed between the attL1 and attL2 recombination sequences. This trigger sequence can be transferred easily to pRISE by an in vitro reaction mediated by LR Clonase, a DNA recombinase that specifically recognizes the attL and attR target sites. Since pRISE carries a pentamer of UASGAL4 in the promoter region, expression of hairpin-shaped dsRNA is dependent on the yeast transcription factor GAL4. Therefore, RNA silencing can be controlled by selecting appropriate ‘driver’ GAL4 transgenes. To ensure efficient transport of the dsRNA to the cytoplasm, we inserted a functional intron between the recombination cassettes. Three versions of pRISE were constructed and each plasmid contains a different intron, derived from fushi-tarazu (ftz), Chitin Synthase 2 (CS-2) or the Ret oncogene. The ftz intron has been studied extensively as a model template for in vitro splicing reactions (Read and Manley, 1992; Rio, 1988), and the CS-2 intron was used in a previously reported RNAi vector for Drosophila (Reichhart et al., 2002; David Gubb, personal communication). The Ret sequence contains two functional introns separated by a small exon and is therefore expected to be spliced effectively (Ryu Ueda, personal communication). Each of these three was integrated between the attR1-ccdB-attR2 cassettes, and the resulting plasmids were designated pRISE-ftz, -CS2 and-Ret, respectively.
 View Details | Fig. 1. Rapid construction of RNAi transgenes using pRISE. (a) Physical map of pRISE-ftz. A target DNA fragment is cloned into the characteristic repeat of the Gateway cassette, resulting in a hairpin-type dsRNA (arrow). Compositions of pRISE-CS2 and pRISE-Ret are exactly same except for the intron sequence between the Gateway cassettes. (b) Schematic representation of RNAi transgene construction using pRISE. A PCR-amplified DNA fragment is first cloned into pENTR-D/TOPO or another appropriate entry vector. Following sequence confirmation, two copies of the insert DNA are integrated into pRISE by in vitro recombination reactions. Recombination between attL and attR sites results in attP and attB sites, respectively. |
Combined with the TOPO cloning system (Invitrogen), plasmid construction was easily achieved by a three-step procedure (Fig. 1b). First, a DNA fragment acting as a trigger sequence was amplified by PCR with a pair of gene-specific primers. Second, the amplified DNA fragment was cloned into the pENTR/D-TOPO entry vector, generating an entry clone. Third, the trigger sequence in the entry clone replaced the ccdB gene of pRISE in an in vitro recombination reaction with LR Clonase. The resulting plasmid, pRISE-dsRNA, contains an inverted repeat of the target sequences and is ready for P-element-mediated transformation in Drosophila. Although the second and third step require conventional transformation into Escherichia coli, the whole procedure can be completed within four days. It is also noteworthy that these processes do not include any restriction enzyme treatment and, therefore, the experimental scheme of plasmid construction is no longer limited by cloning site selection. During plasmid constructions, we found that about half of the resulting plasmids carry the intron sequence in the opposite orientation (Fig. 2), probably due to recombination between inappropriate pairs of att sites (Fig. 2b). The direction of the intron sequence should therefore be confirmed by sequencing or by digestion with appropriate restriction enzymes (Fig. 2c).
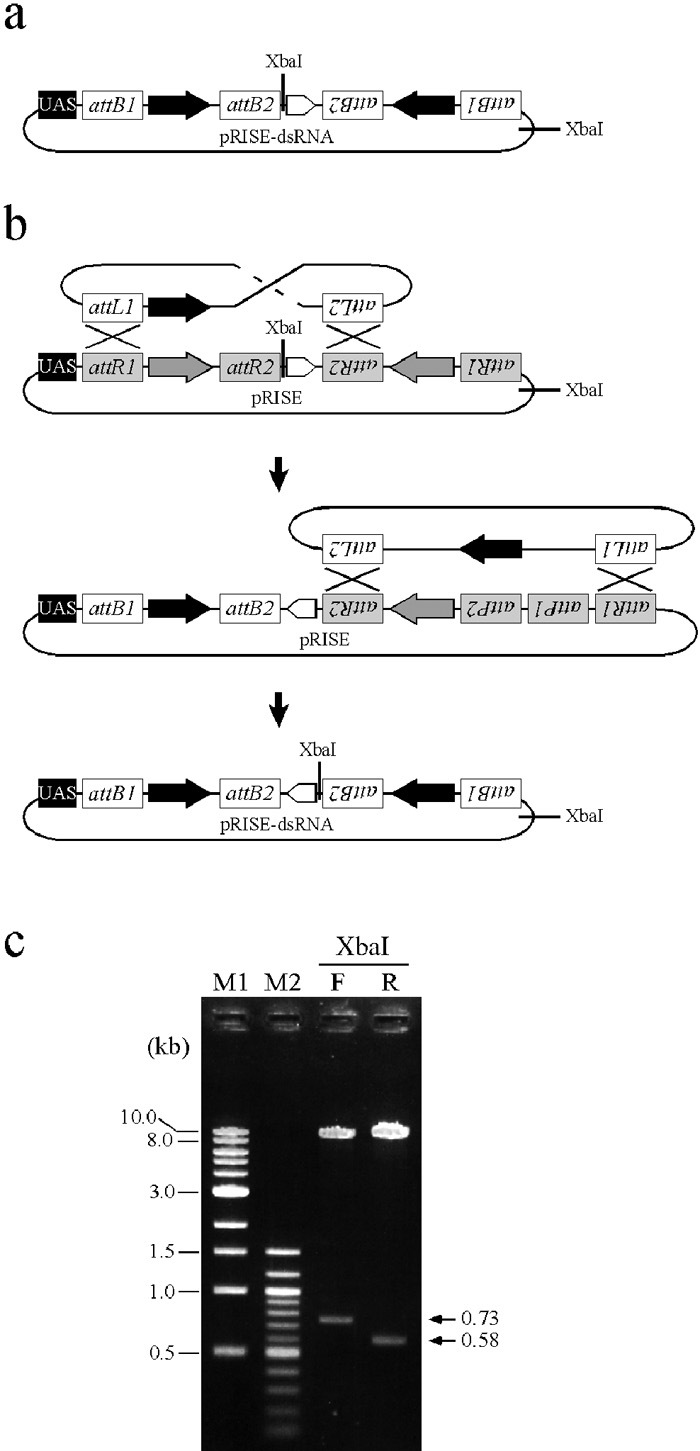 View Details | Fig. 2. Detection of inappropriate recombination during the in vitro reaction. (a) pRISE-dsRNA plasmids contain two XbaI sites: one abuts the functional ftz intron and the other is located outside the Gateway recombination cassettes. (b) An inverted intron sequence in pRISE-dsRNA plasmids can be accounted for by inappropriate recombinations between attL sites of the entry clone and attR sites of pRISE. (c) To determine the orientation of the intron sequence, pRISE-dsRNA plasmids are digested with XbaI. In the case of pRISE-ds-EGFP, 730- and 580-bp fragments (arrowheads) are detected when desired and inappropriate recombination, respectively, has occurred. M1: DNA marker (1kb ladder), M2: DNA marker (100 bp ladder), F: pRISE-ds-EGFP with the ftz intron of forward orientation, R: pRISE-ds-EGFP with the ftz intron of reversed orientation. |
To assess the efficiency of RNAi induction by the pRISE-dsRNA transgenes, we examined reporter gene expression in the presence or absence of the pRISE-dsRNA plasmid in Drosophila S2 cells. As the reporter, we used a UAS-EGFP transgene driven by Actin5C-GAL4, and its fluorescence was easily detected (Fig. 3a, 1st row). When the pRISE-ds-EGFP plasmids were co-transfected with the reporter, fluorescence of EGFP was dramatically reduced (Fig. 3a, 2nd column), while fluorescence of DsRed, also co-transfected as a control, was not affected (Fig. 3a, 3rd column). The effectiveness of RNAi induction was dependent on the intron composition of each vector: the silencing effect of ftz-containing transgenes was similar to that of Ret, whereas the effect of CS-2 was relatively weak. Northern blot analysis showed that accumulation of EGFP mRNA was inhibited in dsRNA-expressing cells (Fig. 3b), to a similar extent as the fluorescence intensity. These results demonstrate that dsRNA produced from pRISE effectively suppresses target gene expression in cultured Drosophila cells. Since the ftz intron is shorter than the Ret intron and does not result in any extra sequence after splicing, further analysis was performed using pRISE-ftz.
 View Details | Fig. 3. pRISE-ds-EGFP induces effective RNAi in Drosophila S2 cells. (a) The UAS-EGFP reporter plasmid and the Act5C-GAL4 expression plasmid were co-transfected in the presence and absence of pRISE-ds-EGFP constructs, and the effects of RNAi were evaluated by EGFP fluorescence. A plasmid encoding DsRed-Express, another fluorescent protein, was also transfected to monitor transfection efficiency. (b) RNA stability was also evaluated by Northern blot analysis. Total RNAs were extracted from the same S2 cells shown in (a). Note that EGFP mRNA levels correlate with those of fluorescence. The ribosomal protein 49 (rp49) gene was used as a loading control. |
We next examined the silencing effect in transgenic animals. To monitor RNAi in individual flies, we chose the white gene as a reporter. The white gene is required for normal pigmentation in the fly eye, and it is easy to see under a standard stereomicroscope when expression of the endogenous white gene is reduced (Lindsley and Zimm, 1992 and references therein). As well as the endogenous white gene, a truncated white gene (referred to as mini-white, which lacks part of the enhancer region and the first intron) is contained in most Drosophila transformation vectors, and also in pRISE-ftz, as a selection marker (Pirrotta, 1988), and the mini-white transgene can thus also be a convenient reporter for evaluation of RNAi effects. A 554-bp cDNA fragment derived from the white ORF was cloned into the pRISE-ftz vector and the resulting pRISE-ds-white plasmid was transformed into flies. When UAS-ds-white transgenic flies were crossed with an Act5C-GAL4 driver strain (Ito et al., 1997), expression of the mini-white transgene was clearly reduced (Fig. 4a, panel 4). These effects were temperature-sensitive, as previously reported in the GAL4-UAS expression system (Duffy, 2002): silencing effects were relatively weak at 18°C, while all flies showed little or no pigmentation at 25°C (data not shown). Consistent with the silencing effect on the mini-white transgene, the endogenous white gene was also repressed in flies carrying both the UAS-ds-white and GAL4 transgenes. In the presence of Act5C-GAL4, the UAS-ds-white transgene effectively reduced eye pigmentation, even though residual pigmentation remained (Fig. 4b, panel 1–2). Another GAL4 driver, photoreceptor cell-specific GMR-GAL4 (Freeman, 1996), also induced RNAi effectively (Fig. 4b, panel 3–4), suggesting that tissue-specific GAL4 drivers are also applicable for RNAi experiments using pRISE-dsRNA transgenes. Taken together, these results demonstrate that the pRISE-dsRNA transgenes can effectively induce RNAi in vivo.
 View Details | Fig. 4. Effective RNAi in transgenic flies using pRISE. (a) Effects of RNAi against mini-white transgenes. All flies shown were females. Genotype of the fly in each panel is (1) y w (the host strain of transgenics) (2) y w; P[w+mC, Act5C-GAL4]25FO1/CyO, Ts(Y:2Lt)B80, Kr– y+ (3) y w; CyO, Ts(Y:2Lt)B80, Kr– y+/+; P[w+mC, UAS-ds-white]4/+ (4) y w; P[w+mC, Act5C-GAL4]25FO1/+; P[w+mC, UAS-ds-white]4/+. (b) Effects of RNAi against the endogenous white gene. All flies were females and carried one copy of the wild-type white gene on the X chromosomes. Genotype of the fly in each panel is (1) y w/+; P[w+mC, Act5C-GAL4] 25FO1/+; TM3, y+ Ser/+ (2) y w/+; P[w+mC, Act5C-GAL4]25FO1/+; P[UAS-ds-white, w+mC]4/+ (3) w/+; P[w+mC, GAL4-ninaE.GMR]12/CyO (4) y w/+; P[w+mC, GAL4-ninaE.GMR]12/+; P[UAS-ds-white, w+mC]4/+. |
Previous studies of RNAi using transgenic flies have employed relatively complex DNA cloning procedures to construct dsRNA transgenes. For example, after the initial PCR cloning, the target sequence had to be inserted into two sites of a transformation vector in separate cloning steps (Lee and Carthew, 2003; Reichhart et al., 2002). In addition, since these methods utilized restriction sites in the transformation vector, addition of extra nucleotides to PCR primers was required. Our results demonstrate that pRISE simplifies these procedures by using an in vitro recombination system. We conclude that pRISE is a useful tool for rapid construction of RNAi transgenes and for functional analysis of genes, especially for those newly identified by recent progress in Drosophila genomics.
We thank Dr. Ian Smith and Dr. Yoshiko Hashimoto for critical reading of the manuscript. We also thank Dr. Ryu Ueda for kindly supplying the Ret intron DNA, Dr. Kunio Inoue (Kobe University) for the ftz intron DNA, and the Kyoto Stock Center for Drosophila strains. This study was supported in part by Grants-in-Aid for the 21st Century Center of Excellence Program, as well as Grants-in-Aid for Scientific Research on Priority Areas, from MEXT to Y.K. This study was also supported by a Grant for Young Scientists from the Foundation for Nara Institute of Science and Technology to T. K., S. I. and Y. K.