| Edited by Toru Terachi. Takanori Ohsako: Corresponding author. E-mail: ohsako@kpu.ac.jp |
The sandy seashore, one of the most severe environments for terrestrial plants, is inhabited by plants that have adapted specifically to this environment. Calystegia soldanella (Convolvulaceae) is a self-incompatible perennial herb that is common in the sandy seashores of the temperate zones of the world. The species proliferates by clonal propagation through the elongation of rhizomes as well as by sexual reproduction. In Japan, C. soldanella is the most common species of sandy coastal vegetation. The wide and abundant distribution of the species suggests that the species possesses a strong ability to disperse and colonize habitat. A high degree of dispersal would result in a genetic admixture and a lack of genetic differentiation among local populations. In Europe, no significant differentiation was found among populations ranging from the Black Sea to the North Sea (Arafeh and Kadereit, 2006). This lack of genetic structure might be due to both seed dispersal by sea currents and high seed longevity (Arafeh and Kadereit, 2006). Allozyme analysis of Korean populations also revealed a low level of genetic differentiation (Chung et al., 1995).
In Japan, C. soldanella is distributed on the shores of Lake Biwa, as well as on the seacoast. The existence of the inland populations provides an opportunity to investigate the effects of strict geographical isolation on the genetic structure of the coastal plants. The milder environmental conditions and the geographical isolation away from the sea should have caused differential directions of natural selection and local genetic drift, respectively. These events might in turn result in the genetic differentiation between the populations on the seacoast and along Lake Biwa. The populations at Lake Biwa differ from those on the seacoast in both flower and seed sizes (Yamada, 1992), suggesting genetic differentiation between them.
To clarify the level of genetic variability and the geographical structure of C. soldanella in Japan, we surveyed nucleotide polymorphisms of the Adh gene that has been the subject of population and evolutionary genetic survey in many plant species (Arabidopsis thaliana: Hanfstingl et al., 1994; Innan et al., 1996; Hordeum vulgare ssp. spontaneum: Cummings and Clegg, 1998; Oryza rufipogon: Yoshida et al., 2004; Yoshida and Miyashita, 2005; Miscanthus sinensis and M. condensatus: Chiang et al., 2003).
Mature plants were collected from populations. Populations investigated in the present study and their geographical locations are shown in Table 1 and Fig. 1. Individuals were sampled at intervals of greater than 10 m to minimize the possibility of sampling multiple ramets belonging to the same genet. Plants were transplanted to 1/5000-a Wagner pots.
 View Details | Table 1 Populations investigated in the present study |
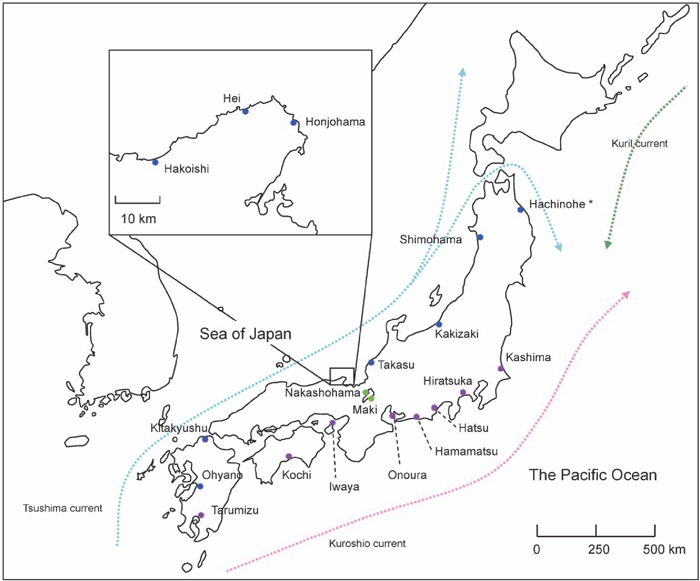 View Details | Fig. 1 The geographical locations of the populations of C. soldanella surveyed in the present study. Populations of the group of Sea of Japan, the group of the Pacific Ocean, and the group of Lake Biwa are indicated with blue, purple, and green circles, respectively. Ocean currents are indicated by broken lines with arrow head. * Hachinohe population was included into the group of Sea of Japan because the location of the population is thought to be related to Tsushima current rather than Kuroshio current. |
Total DNA was extracted from leaves using GenElute Plant Genomic DNA Miniprep kits (Sigma-Aldrich, MO). To isolate a partial region of the Adh gene from C. soldanella, PCRs were performed using the primers PhAdhex2fw (5’-CTGATGTCTATTTCTGGGAAGC-3’) and PhAdhex5rv (5’-CAGCTATTACCTCTTGAACTGG-3’), designed based on the nucleotide sequence of the mRNA of the Adh2 gene of Petunia x hybrida (AY231365). PCRs were performed in a volume of 50 μL containing 0.5 U EX Taq DNA polymerase (TaKaRa, Japan), reaction buffer, 200 μM each dNTP, 500 nM each primer, and ~10 ng total DNA of an individual from Onoura population, Mihama town, Aichi Prefecture (“Onoura 1”). Thermal conditions were as follows: 94°C for 5 min, 30 cycles of 94°C for 15 sec, 52°C for 15 sec, and 72°C for 40 sec, and a final extension of 72°C for 7 min. After purification using Geneclean II (Qbiogene, OH), the PCR fragment (~ 0.7 kb) was cloned into the pGEM-T Easy vector and sequenced using M13 FW (5’-GTAAAACGACGGCCAGTGA-3’) and M13 RV (5’-CAGGAAACAGCTATGACCAT-3’) on a CEQ8800 Genetic Analysis System (Beckman Coulter, CA). Based on the nucleotide sequence of the partial Adh gene of C. soldanella, inverse PCR (i-PCR) (Ochman et al., 1988) was performed to isolate the rest of the gene region. For i-PCR, 150 ng of total DNA of “Onoura 1” was digested with 30 U of HindIII and purified by ethanol precipitation. Digested total DNA was dissolved in 6 μL of dH2O, and self-ligation was induced using Ligation High (Toyobo, Japan) in a total volume of 9 μL. After overnight incubation of the reaction mixture at 16°C, self-ligated DNA was used as the template for the first i-PCR. The first i-PCR was performed in a total volume of 20 μL containing 1 U of EX Taq DNA polymerase, reaction buffer, 200 μM each dNTP, 500 nM each primer [CsADHinverse 5-1 (5’-CGGCCTCGTGTCCGAGGATA-3’) and CsADHinverse 3-1 (5’-GGTTCCAAGCAGATTTGAGCTAGG-3’)], and 2 μL of self-ligated DNA. The second i-PCR was performed in a total volume of 50 μL containing 1 U of EX Taq DNA polymerase, reaction buffer, 200 μM each dNTP, 500 nM each primer [CsADHinverse 5-2 (5’-CAGGATTTTGCCCCTGCAAGAT-3’) and CsADHinverse 3-2 (5’-GGTGTGACTGACTTTGTGAACCC-3’)], and 2.5 μL of a 10–3 dilution of the first i-PCR aliquot. The thermal conditions for the first and second i-PCRs were as follows: denaturing at 94°C for 3 min, 30 cycles of 94°C for 20 sec, 58°C (first) or 56°C (second) for 20 sec, and 72°C for 3 min, and a final extension of 72°C for 5 min. Based on the nucleotide sequence obtained by the i-PCR, primers CsAdhex1fw (5’-GGCAAGTCATTCGATGCAAAG-3’) and CsAdh3’rv (5’-CTGGGAAATGAAAACATGAATGAG-3’) were designed to amplify the entire region of the Adh gene. Using the primer pairs CsAdhex1fw / CsAdh3’rv and PhAdhex2fw / CsAdh3’rv, the Adh gene was amplified in a total volume of 50 μL containing 1 U of high-fidelity Pyrobest DNA polymerase (TaKaRa, Japan), reaction buffer, 200 μM each dNTP, 500 nM each primer, and ~50 ng of total DNA. The thermal conditions were as follows: denaturing at 94°C for 3 min, 30 cycles of 94°C for 15 sec, 56°C for 15 sec, and 72°C for 2 min. PCR fragments were purified using Geneclean II and cloned into the pCR TOPO vector (Invitrogen, CA). The cloning of the fragment amplified with CsAdhex1fw and CsAdh3’rv was unsuccessful after several dozen attempts. On the other hand, the fragment amplified by PhAdhex2fw and CsAdh3’rv could be cloned with high efficiency. The cloned fragment was sequenced on an ABI 3730xl sequencer (Applied Biosystems, CA). A single clone was sequenced for each individual because the possibility of occurrence of PCR artifact has shown to be quietly low with the same experimental procedure in the study on the Als genes of Monochoria vaginalis (Ohsako and Tominaga, 2007). The DNA sequences are available through DDBJ (Accession numbers AB366023–AB366114).
Nucleotide sequences were aligned using ProSeq 2.91 (Filatov, 2002). Nucleotide diversity (π) and Watterson’s θ were estimated using DnaSP 4.0 (Rozas et al., 2003). Estimation of Tajima’s D values (Tajima, 1989) for entire region, noncoding region (intron and 3’ flanking region) and coding region and test for their significance using coalescent simulation were performed with ProSeq 2.91. Genetic differentiation among the three geographically isolated groups of populations, i.e., populations along Sea of Japan (nine populations), those along the Pacific Ocean (eight populations), and those on the shores of Lake Biwa (two populations), was assessed by analysis of molecular variance (AMOVA) (Excoffier et al., 1992) using Arlequin ver. 2.000 (Schneider et al., 2000). The grouping of populations of sea coasts was based on the assumption that gene flow among populations is largely due to sea currents, i.e., Tsushima current at Sea of Japan and Kuroshio current at the Pacific Ocean. According to this idea, Hachinohe population, along the Pacific Ocean, was included into the group of Sea of Japan because the location of the population is thought to be related to Tsushima current rather than Kuroshio current (Fig. 1). Phylogenetic analysis was performed by the NJ method based on the numbers of base substitutions per site estimated by Kimura’s two-parameter method (Kimura, 1980) using PAUP 4.0 (Swofford, 2002).
Although a full-length sequence of the entire coding region of the Adh gene of C. soldanella was not obtained due to an unexpected failure in cloning the fragment, the structure of the gene was inferred based on a contig of the sequences of the partial coding region and the 5’ and 3’ i-PCR fragments. The length of the coding region of the Adh gene of the C. soldanella isolated in the present study was 1889 bp. Based on comparisons with the mRNA sequence of the Adh2 gene of P. hybrida (AY231365.1), the Adh gene of C. soldanella was inferred to include nine exons (1137 bp in total) and eight introns (752 bp). The fragment amplified with the primer pair PhADHex2fw and CsADH3’rv was 1638–1641 bp long and corresponded to a region from the 3’ end of exon 2 to the 3’ noncoding region.
A total of 44 polymorphic sites were detected among 1625 sites without alignment gaps. Out of these sites, 34 were singleton. All of other polymorphisms were shared with multiple populations, i. e., there is no exclusive polymorphism. In addition, length mutations were detected at intron 2 (poly-T repeat) and intron 7 (2-bp insertion and poly-A repeat). Estimates of nucleotide diversity (π) and Watterson’s θ are shown in Table 2. The nucleotide diversity value for the entire region was 0.0021. Nucleotide diversity values for nonsynonymous sites (πnonsyn) were much lower than those for synonymous sites (πsyn) and silent sites (πsilent), suggesting strong functional constraints on the gene. Tajima’s D values were significantly negative for all samples (Table 2). For smaller subsamples, values did not significantly deviate from zero. Tajima’s D values were also estimated for noncoding region (intron and 3’ flanking region) and coding region separately (Table 3). Coalescent simulations with constant population size indicated that D value for coding region was significantly negative but that for noncoding region was not significantly deviated from zero. Separate analyses of synonymous and nonsynonymous substitutions within coding region resulted in significantly negative value for only nonsynonymous substitutions. The neutrality test of McDonald and Kreitman (1991) on nucleotide substitutions in the coding region (Table 4) did not detect natural selection on the gene (G = 0.744; P > 0.10). These results suggest that nonsynonymous mutations are subject to negative purifying selection, whereas other mutations are selectively neutral.
 View Details | Table 2 Nucleotide variability within groups of populations |
 View Details | Table 3 Results of Tajima’s test for neutrality of nucleotide polymorphism |
 View Details | Table 4 Nucleotide substitutions in the coding region of Adh in C. soldanella and its relative, C. hederacea |
AMOVA with two different groupings revealed that the group of populations on the shore of Lake Biwa was not differentiated from those on Sea of Japan and the Pacific Ocean (Table 5). This result is inconsistent with the morphological differentiation shown by Yamada (1992), though his data probably contained environmental variance as well as genetic variances. More than 80% of the total genetic variation was found within the populations. Moreover, the FST values among populations were 0.162 (three groups), 0.128 (two groups), and 0.176 (without grouping). These results indicate a significant but low level of differentiation among populations. Because there is an excess in rare variants in exons, especially for nonsynonymous substitutions as indicated with significantly negative D values, AMOVA was also conducted separately on noncoding region. The results were essentially the same as those on the entire region (Table 5). The phylogenetic relationships among nucleotide sequences inferred with the NJ method are shown in Fig. 2. In the tree, individuals of three geographical groups are interspersed and most of the populations do not form monophyletic groups. These results also indicate a lack of geographical structure in genetic variability.
 View Details | Table 5 Results of AMOVA |
 View Details | Fig. 2 A neighbor-joining tree showing the phylogenetic relationships among nucleotide sequences of the Adh gene of C. soldanella based on the numbers of nucleotide substitutions per site between sequences inferred by Kimura’s two-parameter method. Populations of the group of Sea of Japan, the groups of the Pacific Ocean, and the group of Lake Biwa are indicated in blue, purple, and green, respectively. Numbers above branches indicate bootstrap support percentage over 50% in 5000 replicates. |
The low level of differentiation among population might be partially due to extensive gene flow, largely by means of sea current-mediated seed migration as discussed previously (Arafeh and Kadereit, 2006). This hypothesis is, however, invalid for the explanation of genetic relationship between sea coastal and inland populations because C. soldanella does not have long-distance terrestrial dispersal system. Some anthropogenic causes might be more responsible for the migration from sea coastal populations to inland.
The degree of genetic differentiation depends on the time of isolation and the effective population size throughout the isolation. Another possible explanation for the low level of genetic differentiation is that the inland populations have not been isolated long enough for genetic differentiation. The origin of the populations on the shores of Lake Biwa is unknown. At ~900000 – 500000 BP, the coastline moved close to the southernmost part of Lake Biwa when Osaka Bay extended its area into the Kyoto and Nara Basins (Yokoyama, 2004). Because water drains from the southern edge of Lake Biwa into Osaka Bay, the ancestor of the inland populations might have colonized via the water connection from the seacoast populations. Since the retreat of the coastline at about 500000 BP, inland populations should have been isolated from the sea coastal populations. The results of the present study suggest that the time since isolation is insufficient for the genetic differentiation between them. Analyses of more thorough data regarding nucleotide variability, in combination with reliable estimates of the separation time, would help clarify the biogeographical and demographic history of the species.