| Edited by Etsuko Matsuura. Yoko Satta: Corresponding author. E-mail: satta@soken.ac.jp. Footnotes: GIs, genomic islands; R-M system, restriction modification system; MFRHA, mannose-fucose-resistant hemagglutinin; CcrM, cell-cycle regulated methyltransferase |
Vibrio parahaemolyticus is a γ-proteobacterium which inhabits marine and estuarine environments in the tropical and temperate zones of the world. Virulent strains cause gastroenteritis in humans through the ingestion of seafood contaminated due to raw or insufficient cooking forms. Virulent strains carry either or both of two virulence genes: the tdh gene encoding the thermostable direct hemolysin (TDH), and the trh gene encoding the thermostable direct hemolysin-related hemolysin (TRH) (Nishibuchi and Kaper, 1995). The enteropathogenicity of V. parahaemolyticus mainly results from the enterotoxic activity of TDH and/or TRH (Nishibuchi and Kaper, 1995). Infections by the virulent strains had been limited to local areas before 1995. However, a new pathogenic type of V. parahaemolyticus emerged in Asia in 1995 and then infection by these new-type strains spread rapidly to North and South America, Europe, and Africa (Okuda et al., 1997; Matsumoto et al., 2000; Martinez-Urtaza et al., 2004; Ansaruzzaman et al., 2005; González-Escalona et al., 2005; Cabanillas-Beltrán et al., 2006; Nair et al., 2007). Because the infection by these strains spread across international borders, these strains have been called “pandemic” or “post-1995 pandemic” strains (Matsumoto et al., 2000; Hurley et al., 2006).
Genetic analyses of these strains isolated in various parts of the world after 1995 demonstrated that these strains had the tdh gene, but lacked the trh gene. Experimental analyses revealed that the activity of TDH in these pandemic strains do not differ from that in non-pandemic virulent strains, suggesting the pandemicity should be attributed to other genetic changes in the genome of new strains (Okuda et al., 1997). Further analysis by the arbitrarily primed PCR showed that the DNA fingerprints of these pandemic strains are identical (Okuda et al., 1997). This implied that they are genetically homogeneous, indicating their clonality. This clonality was also supported by the fact that most of these strains belong to a single “new” serotype O3:K6 (Okuda et al., 1997). Additionally, several PCR analyses identified nucleotide sequences unique to these pandemic strains (Matsumoto et al., 2000; Nasu et al., 2000; Iida et al., 2001; Williams et al., 2004; Okura et al., 2004, 2005). However, where and how these pandemic strains have emerged and how the infection by these strains have spread throughout the world are not known to date.
V. parahaemolyticus has two chromosomes (Yamaichi et al., 1999). The whole genome sequencing of RIMD2210633, one of the post-1995 pandemic strains, revealed that chromosome 1 (~3.29 Mb) has most of the essential genes which are required for growth and viability, and chromosome 2 (~1.88 Mb) has genes for adaptation to environmental changes (Makino et al., 2003). Further, recent analyses of the genome sequence identified seven genomic islands (GIs), and four of them are specific to post-1995 pandemic strains (Hurley et al., 2006). GIs are genomic segments that are acquired probably by horizontal gene transfer and these GIs, in general, play important roles in pathogenicity, symbiosis, fitness, metabolic pathways, and antibiotic resistance for pathogenic and environmental bacteria (Dobrindt et al., 2004). Therefore, the four GIs are thought to be potential factors of pandemicity in the V. parahaemolyticus genome. GIs specific to the pandemic strains are VPaI-1 (24 kb: VP0380–VP0403), VPaI-4 (17 kb: VP2131–VP2144), VPaI-5 (12 kb: VP2900–VP2910) and VPaI-6 (27 kb: VPA1253–VPA1270). VPaI-6 locates on chromosome 2, while the other three locate on chromosome 1. However, the function and origin of these GIs have not been fully examined. Among the four GIs, recently VPaI-1 is suggested to be one of causes of pandemicity due to the presence of a virulence-associated gene (Wang et al., 2006). In addition, we found that a mutant of a pandemic strain of which the VPaI-1 region was deleted exhibited reduced swarming ability under a certain culture condition and reduced ability to adapt to cold and heat shocks (Kamruzzaman et al., manuscript in preparation). We therefore became interested in the VPaI-1 region in particular, although other GIs or other specific changes in the pandemic strains may also play important roles in pandemicity.
In this paper, we examine details of the function and origin of VPaI-1 genes by searching the homologs in Bacteria and Archaea and then discuss the roles of VPaI-1 in the pandemicity of V. parahaemolyticus.
The genomic sequences of V. parahaemolyticus RIMD2210633, V. vulnificus CMCP6 and Shewanella sp. MR-7 were retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/) and the DDBJ database (http://gib.genes.nig.ac.jp/).
We examined that the conserved protein domain of 24 genes in the VPaI-1 using hidden Markov models from the Pfam web site (http://pfam.jouy.inra.fr/index.shtml). The presence of homologs of these 24 genes in Bacteria and Archaea was searched using the tblastn program at the NCBI web site (http://www.ncbi.nlm.nih.gov/BLAST/). The tblastn program can be used to infer functional and evolutionary relationships between sequences by comparing protein sequences to translated nucleotide sequence databases and calculating the statistical significance of matches. The significance of results is measured by e-values. We set the criterion of e-values to be 1.0e-3. We excluded genes or regions even with significant e-values when they showed low bitscores (50), because low scores indicate that the similarity is limited to a part of genes or regions.
Nucleotide or amino acid sequences were aligned using the program CLUSTALW (http://align.genome.jp/). The number of nucleotide or amino acid differences per site (p-distances) between homologous genes was calculated using the Mega 3.1 software program (Kumar et al., 2004). Nucleotide diversities or divergences at synonymous sites were estimated with the Nei-Gojobori method (p-distance) by using the MEGA 3.1 software.
To research the syntenic regions between different bacterial genomes, we used the dotter program (http://www.cgb.ki.se/cgb/groups/sonnhammer/Dotter.html).
For the phylogenetic analysis of 16S rRNA, nucleotide sequences from 26 bacterial strains were retrieved from NCBI database. Most of these strains are closely related γ-proteobacteria species to V. parahaemolyticus. Gene IDs of these sequences are shown in a supplementary Table (Table 1). Phylogenies were constructed by Neighbor Joining (NJ), Maximum Parsimony (MP), and Minimum Evolution (ME) methods implemented in the Mega 3.1 software.
 View Details | Table 1 16S rRNA sequences from 26 bacterial strains |
The whole genome sequence of V. parahaemolyticus RIMD2210633 (http://www.ncbi.nlm.nih.gov/sites/entrez) shows that the VPaI-1 locates on chromosome 1 (381054–403433) and contains 24 genes (Table 2). As reported by Wang et al. (2006), Pfam analysis shows that of these 24 genes, nine (VP0380, VP0386, VP0387, VP0388, VP0394, VP0395, VP0399, VP0400 and VP0402) encode proteins with known function (Table 2). Three genes (VP0387, VP0388 and VP0395) encode type I restriction modification system genes (R-M system: Hurley et al., 2006; Wang et al., 2006). The function of the R-M system is considered as protecting the bacterium from viral infections (Boyer, 1964; Uetake et al., 1964; Lederberg, 1965; Arber, 1974; Bickle and Kruger, 1993; Casadesús and Low, 2006). In addition, VP0402 has a conserved HipA-like domain that may act against antibiotic activity (Falla and Chopra, 1998), and VP0394 encodes DNA methylase which may play an important role in virulence (Wang et al., 2006). The other four encode integrase (VP0380), inner membrane protein (VP0386), transcriptional regulator (VP0399), and transmembrane protein (VP0400), respectively.
 View Details | Table 2 Pfam and tblastn search of VPaI-1 embedded 24 genes |
The remaining 15 genes (VP0381, VP0382, VP0383, VP0384, VP0385, VP0389, VP0390, VP0391, VP0392, VP0393, VP0396, VP0397, VP0398, VP0401 and VP0403) encoded “hypothetical proteins” (Table 2). In order to infer functions of these proteins, we examined similarity with functional motifs in the Pfam database. One gene (VP0382) shows the sequence similarity to “Calcineurin-like phosphoesterase”, but the other 14 genes do not show any similarity to known functional motifs (Table 2).
To infer the origins of these 24 genes, we examined whether homologs of these genes exist in Bacteria and Archaea using tblastn (Table 2). Five hypothetical protein genes (VP0381, VP0384, VP0385, VP0396 and VP0397) are found in V. parahaemolyticus only and thus this analysis does not give any clues for their origins. The other seven genes (VP0380, VP0387, VP0388, VP0394, VP0395, VP0400 and VP0402) are present in 73 to over 100 strains of proteobacteria (α, β, γ, δ, ε), other phyla of Bacteria and Archaea, suggesting a relatively wide distribution and ancient origin of genes. Functions of these seven genes are related to DNA modifications, virulence, and antibiotic activity. The remaining 12 genes show sequence similarities with a limited number of strains (2–26 strains). In particular, seven (VP0389, VP0390, VP0391, VP0392, VP0393, VP0398 and VP0399) are distributed in two to seven γ-proteobacteria strains only including V. parahaemolyticus, V. vulnificus, V. fischeri, V. cholerae and Shewanella sp. Except for VP0399, which encodes a putative transcriptional regulator, encoding products were annotated as hypothetical proteins.
To examine whether presence or absence of genes are related to phylogenetic relationships of bacteria, the NJ tree based on 16S rRNA were constructed (Fig. 1). The topology does not change when a tree was constructed by using MP or ME method. Then the presence or absence of homologs of genes was examined along the phylogeny. The R-M system genes (VP0387, VP0388 and VP0395) are widely distributed within proteobacteria and other phyla of Bacteria and Archaea (Table 2, Fig. 1). The three genes of the R-M system are tandemly located on genomes in many species, such as Escherichia coli O157:H7 str. Sakai (locus tag: ECs5306–ECs5307–ECs5308), and Photobacterium profundum SS9 (locus tag: PBPRA1802–PBPRA1803–PBPRA1804). However, the arrangement of the R-M system genes in the VPaI-1 is different from that in other species. There is an insertion of six genes (VP0389 to VP0394) in the R-M system (Fig. 2b). Among the six genes, VP0394 shows a wide distribution over many species as mentioned above (Table 2, Fig. 1), while VP0393 is limited to V. cholerae and V. parahaemolyticus. Two (VP0389 and VP0391) of the remaining four genes are found in five and seven γ-proteobacteria strains, respectively (Table 2, Fig. 1), including V. vulnificus CMCP6 and Shewanella sp. MR-7, but VP0390 and VP0392 are found in the V. vulnificus CMCP6 and Shewanella sp. MR-7 only. Of the six inserted genes, four (VP0389, VP0390, VP0391 and VP0392) are shared with V. vulnificus CMCP6 and Shewanella sp. MR-7 strains (Fig. 1).
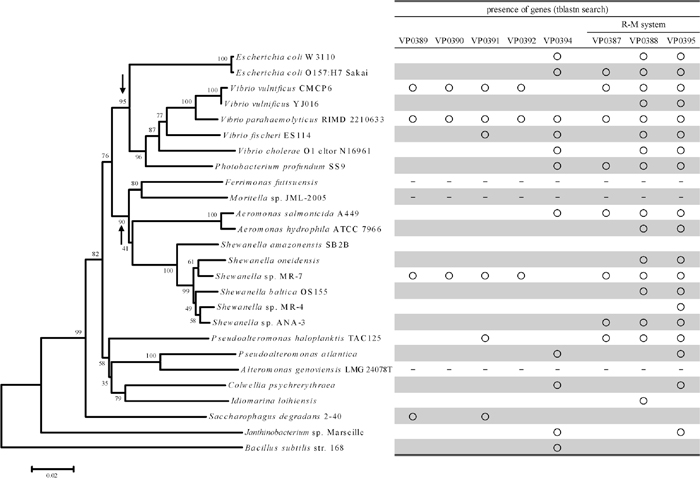 View Details | Fig. 1 The NJ tree based on 16S rRNA nucleotide sequences. Nucleotide sequences were from 26 bacterial strains (see Materials and Methods). The presence (circle) of homologs VP0389, VP0390, VP0391, VP0392, VP0394, and three R-M system genes are represented in a table on the right hand side of the tree. The scale bar (the number of nucleotide differences per site) is indicated at the bottom. Each black arrow indicates a cluster containing either Shewanellas or Vibrios. The symbol of “–” shows that presence or absence of homologs could not be judged because whole genome sequences are not determined. |
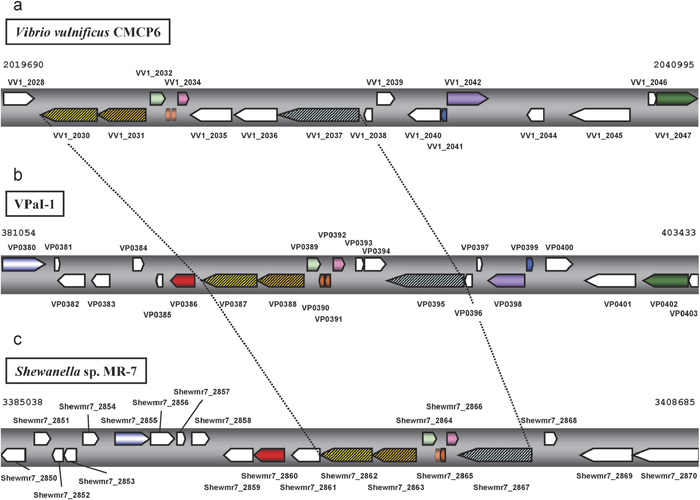 View Details | Fig. 2 Comparison of syntenic region between VPaI-1 and other bacterial genomes. (a): 21.3 kb region of V. vulnificus CMCP6 on chromosome 1 (2019690–2040995), (b): 22.3 kb region of V. parahaemolyticus RIMD2210633 in VPaI-1 on chromosome 1 (381054–403433), (c): 23.6 kb region of Shewanella sp. MR-7 on chromosome (3385038–3408685). Pentagon arrows represent annotated genes on whole genome databases, and annotated locus tags were shown above or below the arrows. Arrows without the tag indicates that ORF was detected in this study. Open pentagon arrows represent that there are no homolog in compared regions among three species. Pentagon arrows in the same color represent that homolog in the region among three species. Shaded pentagon arrows indicate a subset of R-M system genes. Broken lines show the syntenic region among three species. |
We used the dotter program in order to compare the sequence and structural similarity of the region containing the four genes and R-M system genes shared by V. parahaemolyticus, V. vulnificus, and Shewanella sp. Comparisons were made between VPaI-1 in V. parahaemolyticus and the corresponding region in V. vulnificus (Fig. 3a) as well as between VPaI-1 and the corresponding region in Shewanella sp. (Fig. 3b). The result showed that, in both cases, there were quite clear homology in a middle of VPaI-1 and that the region contains exactly the four genes and the three R-M system genes (Fig. 2a, 2b, 2c), suggesting the synteny of the region (the syntenic region). Since the syntenic region is observed only the three species (Fig. 2, Fig. 3), this region including four genes (VP0389, VP0390, VP0391 and VP0392) in V. parahaemolyticus shares the common ancestor with Shewanella sp. MR-7.
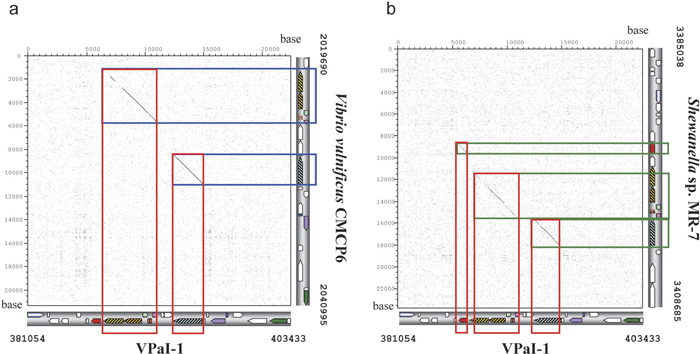 View Details | Fig. 3 Dotplot analysis of VPaI-1 region vs. the syntenic region in other bacteria. The horizontal axis show VPaI-1 on the chromosome 1 in V. parahaemolyticus RIMD2210633 (381054–403433). Open box (red, blue, and green) shows syntenic region of the three species. The location of genes in each genome is shown bottom or right side of the plot. (a): VPaI-1 vs. 21.3 kb region of V. vulnificus CMCP6 on chromosome 1 (2019690–2040995) (vertical axis). (b): VPaI-1 vs. 23.6 kb region of Shewanella sp. MR-7 on chromosome (3385038–3408685) (vertical axis). |
Searches with tblastn reveal that some homologs of the 24 VPaI-1 genes are relatively widely spread, while others are limited to a few species. This observation indicates that the VPaI-1 does not evolve as a unit, although the VPaI-1 region has direct repeat sequences (47 bp) at the both ends (Hurley et al., 2006; Wang et al., 2006) and the region seems to be inserted in the V. parahaemolyticus genome as a single unit. Among the 24 genes, three R-M system genes are shared with many strains (Fig. 1, Table 2). However, in V. parahaemolyticus, V. vulnificus, and Shewanella sp., four genes are inserted in the R-M system gene region and the region shows the synteny among the three species. Interestingly, these four genes are not observed in different strains or closely related species. Pandemic strains of V. parahaemolyticus possess the region but non-pandemic strains do not (Hurley et al., 2006). Similarly the presence or absence of the syntenic region is observed between V. vulnificus CMCP6 and YJ016. Nineteen genomic islands in CMCP6 are reported (van Passel et al., 2005) and five (VVI-1–VVI-5) are found to be specific to CMCP6 compared with YJ016 (Quirke et al., 2006). Among the five GIs, VVI-2 includes the syntenic region (Fig. 2a, 2b, Fig. 3a). In Shewanella sp., a strain MR-7 possesses the region, whereas other Shewanella species (S. amazonensis, S. oneidensis, S. baltica OS155, S. sp. MR-4 and S. sp. ANA-3) do not (Fig. 1). In addition, the phylogeny based on 16S rRNA genes (Fig. 1) shows that a cluster containing V. parahaemolyticus and V. vulnificus is different from a cluster containing several Shewanella strains. The topology is supported by relatively high bootstrap probabilities (95% and 90%, respectively) in not only the NJ tree but also MP or ME trees (Fig. 1). The presence or absence of the four genes (VP0389, VP0390, VP0391 and VP0392) appears discontinuous on the phylogeny (Fig. 1). These observations suggest that the independent and frequent gain or loss of these genes has taken place in the evolutionary course of these bacteria.
In order to know the origin of the VPaI-1, the next question is when the acquisition (probably by horizontal gene transfer) of the above syntenic region occurred. If this has occurred from Shewanella sp. directly and recently or just before epidemiological appearance of pandemic strains of V. parahaemolyticus, the nucleotide sequences of the region should be identical or nearly so between the two species. To test this hypothesis, we examined the nucleotide sequence identities in the syntenic region of the two species. The corresponding region in V. parahaemolyticus (VP0387–VP0395: 8420 bp) shows 63% identity with that of Shewanella sp. (Shewmr7_2862–Shewmr7_2867: 7177 bp), suggesting that the recent acquisition of the region from Shewanella sp. is unlikely. Further, the average number of differences per site in amino acid sequences over the four pairs of homologous genes (VP0389–VP0392) in the syntenic region are 10% for V. parahaemolyticus and V. vulnificus, 47% for V. parahaemolyticus and Shewanella sp., and 46% for V. vulnificus and Shewanella sp. (Table 3). These values seem to indicate that on average V. parahaemolyticus and V. vulnificus are more closely related to each other than to Shewanella sp. Divergences at synonymous sites of genes in the syntenic region except for VP0387 also support this phylogenetic relationship (Table 4). Genes except for VP0387 show 0.108 to 0.325 of synonymous divergence between two Vibrio species while those between Shewanella and Vibrio are much larger (0.564 to 0.816) (Table 4). The synonymous divergence between two Vibrios at VP0387 in the R-M system, however, is different from corresponding divergences at other loci. The former divergence is as large as that between Shewanella and Vibrio. Further, since the synonymous divergence between Shewanella and Vibrio at VP0387 does not differ from values at other loci, this may suggest independent horizontal transfer of VP0387 homolog to these three species from unknown origins. But in this case, donors of the VP0387 homolog should be equally distantly related to each other. On the other hand, the synonymous divergence between two Vibrios at other loci is similar to each other, although face values appear different due to large standard errors because of the small number of synonymous sites of each gene (Table 4).
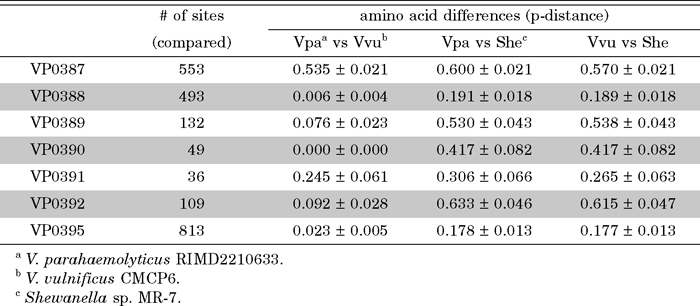 View Details | Table 3 The number of amino acid differences per site (p-distances) in the seven genes |
 View Details | Table 4 The comparison of synonymous divergences at seven genes in the syntenic region |
In V. parahaemolyticus, it is well known that there are two types of cells, translucent (TR) and opaque (OR), of which the former has a swarming ability (Jaques and McCarter, 2006) and that the swarming ability is one of characteristics for pandemic strains (Yeung et al., 2002). Although recently three regulators of the swarming ability have been reported, results suggest the presence of additional factors of the swarming regulation in V. parahaemolyticus (Jaques and McCarter, 2006).
VP0394 is annotated as “hemagglutinin associated protein” in NCBI database. Blast search (tblastn) showed that the homolog of VP0394 is found in not only proteobacteria (α, β, γ, δ, ε), but also other phyla and domain (Firmicutes: Streptococcus pneumoniae D39, Archaea: Thermoplasma acidophilum, etc.) (Table 2), indicating a wide distribution of VP0394 homologs. Wang et al. (2006) suggested that VP0394 is a homolog of a mannose-fucose-resistant hemagglutinin (MFRHA) and a cell-cycle regulated methyltransferase (CcrM). The defective mutant of MFRHA in V. cholerae exhibited attenuation in virulence potential through colonization in an infant mouse (Franzon et al., 1993). On the other hand, the function of CcrM is known as an “orphan” DNA methyltransferase (Stephens et al., 1996; Wright et al., 1997; Casadesús and Low, 2006). Further, CcrM in an α-proteobacterium, Caulobacter crescentus, play an important role for cell-type determination mediated by expression of class II flagellar genes. C. crescentus generates two distinct cell types, a motile swarmer cell and a stalked cell (Stephens et al., 1995; Marczynski and Shapiro, 2002). The expression level of CcrM protein controls the amount of transcripts of class II flagellar genes, thereby being related to the development of swarmer cells (Stephens et al., 1996; Wu et al., 1998; Reisenauer et al., 1999). Taken together, functions of the two proteins (MFRHA and CcrM) suggested that VP0394 may play a potential role in both the swarming regulation and virulence in post-1995 V. parahaemolyticus pandemic strains.
V. vulnificus seems to carry the gene associated with swarming (Kim et al., 2006; Kim et al., 2007). However, it is noted that VP0394 was not found in the closely related two V. vulnificus strains (CMCP6 and YJ016) using blastp search (data not shown). This might be consistent with implications of a unique controlling system of swarming ability in V. parahaemolyticus (Jaques and McCarter, 2006).
Nishina et al. (2004) reported that pandemic strains are able to grow better at low temperature (15°C) than non-pandemic strains suggesting the presence of temperature resistance-related genes in V. parahaemolyticus.
Genus Shewanella is known to be a cold-adapted marine bacterium, and many of the Shewanella species can grow at 4°C (Bowman, 2005). Shewanella sp. is closely related to S. oneidensis (Venkateswaran et al., 1999), a well-known piezophile bacterium which can grow at between 4 and 40°C (Bowman, 2005). These results imply the possibility that the acquisition of the syntenic region may give rise to some kind of resistance to temperature stress in post-1995 pandemic V. parahaemolyticus strains. In particular, the limited presence of VP0390 and VP0392 in the V. vulnificus CMCP6 and Shewanella sp. MR-7 (Fig. 1) suggests that these two might be candidates of putative cold adaptation genes. In fact, V. vulnificus in general are able to adapt to cold temperatures (Bang and Drake, 2002; Bryan et al., 1999). Therefore the cold adaptation of V. vulnificus can be also related to the presence of this syntenic region. However, in order to confirm the responsibility of the two genes for the cold adaptation in post-1995 pandemic V. parahaemolyticus strains, further gain and loss of function experiments are necessary.
We hypothesize that cold adaptation plays an important role in the post-1995 pandemicity of V. parahaemolyticus strains. The preexisting pathogenic strain of V. parahaemolyticus with the tdh gene has gained putative cold adaptation genes probably by a horizontal gene transfer, and consequently the strain shows high survivability at low temperatures or multiplication in cold environment. Thus, this strain can better survive in refrigerated or frozen seafood during export/import. Improvements in freezing technology may have been a selective pressure for the emergence of the new V. parahaemolyticus strain. If this cold or frozen seafood possessing new strains are shipped to global markets and were ingested without adequate cooking, resulting in the seafood-borne illness, it can explain at least in part the worldwide spread of infection by the pandemic strains. We are currently examining this hypothesis.
We thank Dr. Naoyuki Takahata for his critical reading of manuscript and for his numerous suggestions. We also thank Mr. Michael Kryshak for his editorial assistance. This work was supported in part by Grant-in-Aid for Scientific Research (191010) from Japan Society for the Promotion of Science to MN; and a grant-in-aid of Ministry of Health, Labor and Welfare, Japan (H17-Sinkou-ippan-019) to MN.
|