| Edited by Hiroshi Nojima. Hidenobu Soejima: Corresponding author. E-mail: soejimah@med.saga-u.ac.jp |
Epigenetic regulation of gene expression plays a critical role in development and differentiation, X inactivation, and genomic imprinting; it also plays a role in several human diseases, including cancer (Jaenisch and Bird, 2003). In human cancers, a number of genes are known to be repressed by epigenetic means, for example aberrant hypermethylation of the promoter CpG island (CGI). One way in which DNA hypermethylation represses gene expression may be inhibition of the binding of transcription factors to their target sequences. However, it is also widely accepted that methylated CpGs are recognized by the methyl-CpG binding proteins, including MeCP2, MBD1, MBD2, MBD3, MBD4 and Kaiso, and that these proteins recruit protein complexes related to histone modification and chromatin remodeling (Jaenisch and Bird, 2003; Bienvenu and Chelly, 2006). Recent studies have shown that of these, only MeCP2, MBD1 and MBD2 act as methyl-CpG binding proteins in mammals (Lopez-Serra et al., 2006).
MeCP2 is a member of the methyl-CpG binding domain (MBD) family and plays a pivotal role in DNA methylation-associated gene repression. MECP2 is a causative gene for Rett syndrome, a dominant X-linked neurodevelopmental disorder in which affected individuals are usually heterozygous for a de novo mutation in MECP2 (Bienvenu and Chelly, 2006). The canonical gene repression function of MeCP2 involves the molecule binding to methylated CpG sites via a conserved MBD, leading to transcriptional repression, which occurs due to recruitment of Sin3A and histone deacetylases (HDACs) and/or mediation of the methylation of histone H3 lysine 9 (Bienvenu and Chelly, 2006; Fuks et al., 2003a; Fuks et al., 2003b). In cell-free system, MeCP2 can bind to unmethylated nucleosomal arrays and repress transcription from both methylated and unmethylated naked DNA (Meehan et al., 1992; Nan et al., 1997; Kaludov and Wolffe, 2000; Georgel et al., 2003). This naturally suggests that MeCP2 may be able to repress gene expression without DNA methylation in living cells; however, this hypothesis has not been tested. Given that mutations in MECP2 cause Rett syndrome, attempts have been made to identify the target genes of MeCP2 in the brain, with the result that a limited number of target genes have been identified (Bienvenu and Chelly, 2006). However, no attempt has been made to perform a genome-wide screen for MeCP2 target genes in cancer cells, and it remains unknown whether MeCP2 regulates gene expression without DNA methylation in living cells. Thus, in this study we screened potential MeCP2 target genes using knockdown (KD) with siRNA and microarray gene expression analyses, and investigated DNA methylation and the binding of MeCP2 to CGIs within promoter regions. We also performed gene ontology (GO) analysis for the identified MeCP2 target genes in an effort to determine if they have any features in common.
LU65 cells derived from human lung cancer and BEAS-2B cells from human bronchial epithelium were cultured in RPMI-1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10% FCS (Gibco, Invitrogen, Carlsbad, CA, USA) and in MEM-α medium (Gibco) supplemented with 10% FCS, respectively. Cells were harvested at 70% confluence.
We transfected LU65 cells with two different dsRNA sets using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The dsRNA sets were as follows: Set A, synthesized dsRNA with sequences 5’-CCUAAUGAUUUUGACUUCACGGUACAG-3’ and 5’-UUACCGUGAAGUCAAAAUCAUUAGGAU-3’ (Hokkaido System Science, Sapporo, Japan); Set B, dsRNAs (Stealth Select RNAi) of three different sequences (Invitrogen, Catalog #1299003). A single nucleotide at the 5’ terminal of the Set A antisense sequence was intentionally mismatched to improve the knockdown effect of the siRNA (Schwarz et al., 2003). At 72 hours after transfection, cells were harvested for use in further experiments.
Total RNA was extracted using an RNeasy mini kit (Qiagen, Hilden, Germany) with an RNase-free DNase kit (Qiagen). Total RNA (500 ng) was reverse-transcribed with random primers using ReverTra Ace reverse transcriptase (Toyobo, Osaka, Japan). Gene expression was quantitated by real-time PCR on an ABI Prism 7000 with TaqMan probe (Applied Biosystems, Foster City, CA, USA) and QuantiTect SYBR Green PCR kits (Qiagen) for MBDs and other genes, respectively, as shown in Table 1. The expression level of each gene was normalized against that of the housekeeping genes GAPDH or β-actin. All quantitative RT-PCRs were performed in triplicate.
 View Details | Table 1 Primers for quantitative RT-PCRs |
Proteins were extracted from LU65 cells with and without MECP2 KD using SDS lysis buffer (2% SDS, 50 mM Tris-HCl, pH 7.5). Aliquots (5 μg) of protein were loaded onto 10% SDS-PAGE gels and electrophoresed, then blotted onto PVDF membranes using a semi-dry blotting method. The membranes were probed with antibodies against MeCP2 (Kudo, 1998), MBD1 (Abcam, Cambridge, UK, cat. 3753 or 2846), MBD2/3 (Millipore, Billerica, MA, USA, cat. 07-199), and β-actin (Sigma, cat. A5441). An ECL plus western blotting detection system (GE Healthcare, Buckinghamshire, UK) was used for detection.
Total RNA (500 ng) from LU65 cells with and without MECP2 KD was amplified using a Low RNA Fluorescent Linear Amplification Kit (Agilent Technologies, Santa Clara, CA, USA) and labeled with Cy-5 and Cy-3, respectively. Labeled cRNAs were co-hybridized with the Whole Human Genome Oligo Microarray (Agilent Technologies), which includes 41,000 human genes and transcripts. Data were extracted from the resulting images using Agilent’s Feature Extraction Software (Agilent Technologies). Microarray data were deposited in a public database, CIBEX (http://cibex.nig.ac.jp/) (accession no. CBX52).
Genomic DNA extracted from LU65 cells with and without MECP2 KD was subjected to sodium bisulfite modification with an EpiTect bisulfite kit (Qiagen). Modified DNA was amplified by PCR with a primer set specific to each gene, followed by cloning and sequencing. DNA methylation status in LU65 and BEAS-2B cells was also analyzed by combined bisulfite restriction analysis (COBRA). All primers used in this study are shown in Table 2.
 View Details | Table 2 Primers and PCR conditions for methylation analyses and ChIP assay |
Chromatin immunoprecipitation for MeCP2 was performed as previously described (Nakagawachi et al., 2003; Higashimoto et al., 2003). Because the number of cells obtained from the siRNA transfection experiment was small, we independently repeated the ChIP assay five times and defined MeCP2 binding as being positive when at least three positive bands were obtained by ChIP-PCR. We defined MeCP2 binding as being negative when ChIP-PCR revealed that at least three bands were absent relative to the input. To quantitatively evaluate the ChIP-PCR products, we used a γ-32P-labeled forward primer and measured the band intensities of the PCR products with a BAS 2000 Bioimaging Analyzer (Fujifilm, Tokyo, Japan).
Gene ontology (GO) analysis was performed using the curated gene annotation database of World Fusion Co. Ltd (Tokyo, Japan). This database contains genes with GO terms that are based on data in the Gene Ontology Database (http://www.geneontology.org/index.shtml) and NCBI database (http://www.ncbi.nlm.nih.gov/), in particular Entrez Gene (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene). As of May 2007, 17,472 human genes were annotated with GO information.
Differences in GO or GO terms between the genes up-regulated by MECP2 KD and all genes in the human genome were assessed using the chi-squared test or Fisher’s exact test. Two-sided probability values of < 0.01 were considered statistically significant.
Quantitative RT-PCR following MECP2 KD in LU65 cells using two siRNA sets revealed approximately 80% reductions in MECP2 expression using both Set A and Set B, relative to control LU65 cells without siRNA (Fig. 1a). Western blotting also revealed approximately 70% reductions in MeCP2 protein expression relative to control LU65 cells using Set A (Fig. 1b). Furthermore, immunostaining revealed that KD had considerably reduced MeCP2 expression in the nuclei (data not shown). Investigation of the mRNA and protein expression levels of MBD1, MBD2, and MBD3 by quantitative RT-PCR and western blotting, respectively, showed no reduction in the expression of any of these MBDs, indicating that our KD process was specific for MECP2, and did not affect other members of the MBD family (Fig. 1b and 1c).
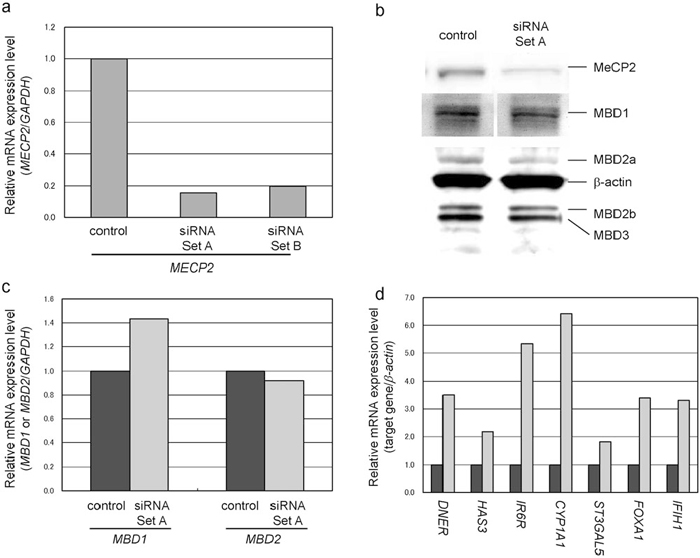 View Details | Fig. 1 MECP2 knockdown. (a) Quantitative RT-PCR of MECP2. Both siRNA sets effectively reduced the mRNA expression level of MECP2. Expression of MECP2 was normalized to that of the housekeeping gene GAPDH. (b) Western blots of the gene products of members of the MBD family. Western blots showed a reduction in MeCP2 but no change in the protein expression of other MBD proteins. Protein expression was normalized to that of β-actin. (c) Quantitative RT-PCR of MBD1 and MBD2. Quantitative RT-PCR showed that MECP2 KD did not reduce the mRNA expression of MBD1 or MBD2. mRNA expression of MBDs was normalized to that of the housekeeping gene GAPDH. The western blotting and quantitative RT-PCR data indicate the specificity of MECP2 KD. (d) Quantitative RT-PCR of the seven selected genes. The expression of all genes was increased by MECP2 KD. Expression of the genes was normalized to that of β-actin. Dark gray bar: control; light gray bar: MECP2 KD. |
With the aim of screening for genes repressed by MeCP2, two independent microarray gene expression analyses using RNAs from two independent MECP2 KD experiments revealed a more than three-fold increase in the expression of 219 and 190 transcripts for siRNA Set A and Set B, respectively. To reduce false-positives, we selected transcripts overlapped between the two experiments. A total of 59 transcripts, including 49 annotated genes, as shown in Table 3, were identified by both siRNA sets. Since a CGI covering the transcription initiation site (promoter CGI) is usually important for gene repression due to DNA methylation (Ushijima, 2005), we searched for the locations of CGIs in each gene using CpG Island Searcher (http://www.cpgislands.com/), applying the criteria of Takai and Jones (Takai and Jones, 2002). Unexpectedly, we found that only half (23 genes) of the 49 genes possessed promoter CGIs. Two other genes had CGIs approximately 200 bp upstream, but not covering, the transcription initiation site. The other 24 genes did not have a CGI in the region from –5 kb to +1 kb relative to the transcription initiation site. None of the 49 up-regulated genes were typical tumor suppressor genes, so we selected 6 genes with promoter CGIs and one with a CGI just upstream of the transcription initiation site for further analysis. These genes are associated with cell growth, differentiation, or transformation. Up-regulation of the seven genes was also confirmed by quantitative RT-PCR (Fig. 1d).
 View Details | Table 3 List of annotated genes with increased expression after MeCP2 knockdown |
Since a core region within a promoter CGI is usually important for gene repression due to DNA methylation (Ushijima, 2005), we performed bisulfite sequencing of regions surrounding the initiation site for the seven selected genes (Fig. 2). Our primer sets for bisulfite sequencing mostly covered the entire CGI region, including the core region. Unexpectedly, the CGIs of five of the analyzed genes were mostly unmethylated before MECP2 KD. Only two genes (DNER and HAS3) harbored methylated CGIs: DNER was densely hypermethylated, whereas HAS3 was approximately half methylated. Further, MECP2 KD did not change the methylation status of any CGI, and these results were confirmed by COBRA (Fig. 3). Our findings indicate that MeCP2 has no effect on DNA methylation. Further examination of the methylation status of all CGIs in BEAS-2B bronchial epithelium cells using COBRA showed that no CGIs were methylated, suggesting that DNER and HAS3 were specifically methylated in LU65 cancer cells (Fig. 3).
 View Details | Fig. 2 Bisulfite sequences of the seven selected genes. Unexpectedly, only two, DNER and HAS3, were methylated on the promoter CGI. The other five genes were mostly unmethylated. Methylation status was not changed by MECP2 KD, indicating that MeCP2 has no effect on DNA methylation. Black circles: methylated CpGs; white circles: unmethylated CpGs. Arrows in the upper and lower rows indicate the regions that were amplified for ChIP-PCR (ChIP) and bisulfite-PCR (BS) for sequencing and COBRA, respectively. Gray bars indicate CpG islands (CGI). Broken arrows indicate transcription initiation sites. |
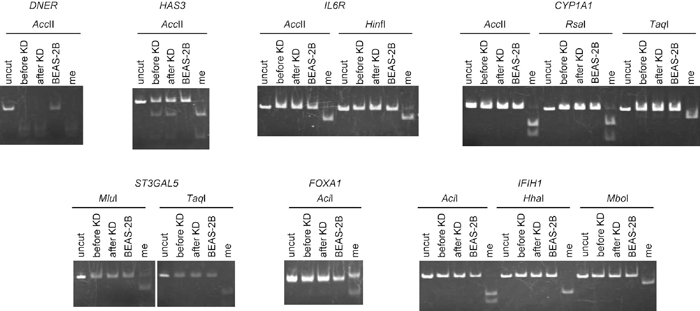 View Details | Fig. 3 Results of COBRA. In LU65 cancer cells, the promoter CGIs of DNER and HAS3 were methylated irrespective of MECP2 KD, which was consistent with the results of bisulfite sequencing. However, in BEAS-2B bronchial epithelium cells, methylation was not observed, suggesting cancer-specific methylation of DNER and HAS3. The CGI of IL6R was unmethylated in both LU65 and BEAS-2B cells. All other genes were unmethylated in both BEAS-2B and LU65 cells. me: control methylated DNA. |
Using five independent ChIP-PCR assays, we determined whether MeCP2 bound to the CGIs of the seven selected genes, as described in the Materials and Methods. Before KD, we found that five (DNER, HAS3, IL6R, CYP1A, and ST3GAL5) of the seven genes were bound by MeCP2, while two (FOXA1 and IFIH1) were not (Fig. 4). For the five CGIs that bound to MeCP2, after MECP2 KD, three genes (DNER, HAS3, and IL6R) had reduced MeCP2 binding, while contradictory or inconsistent results were obtained for two (CYP1A and ST3GAL5), over five independent experiments. We speculated that specific chromatin conformation of the two genes might influence ChIP procedure, leading to such contradictory or inconsistent results. In summary, MeCP2 constitutively bound to the CGIs of five of the seven selected genes before KD, three of which showed reduced MeCP2 binding after KD. Two of the selected genes were not bound by MeCP2.
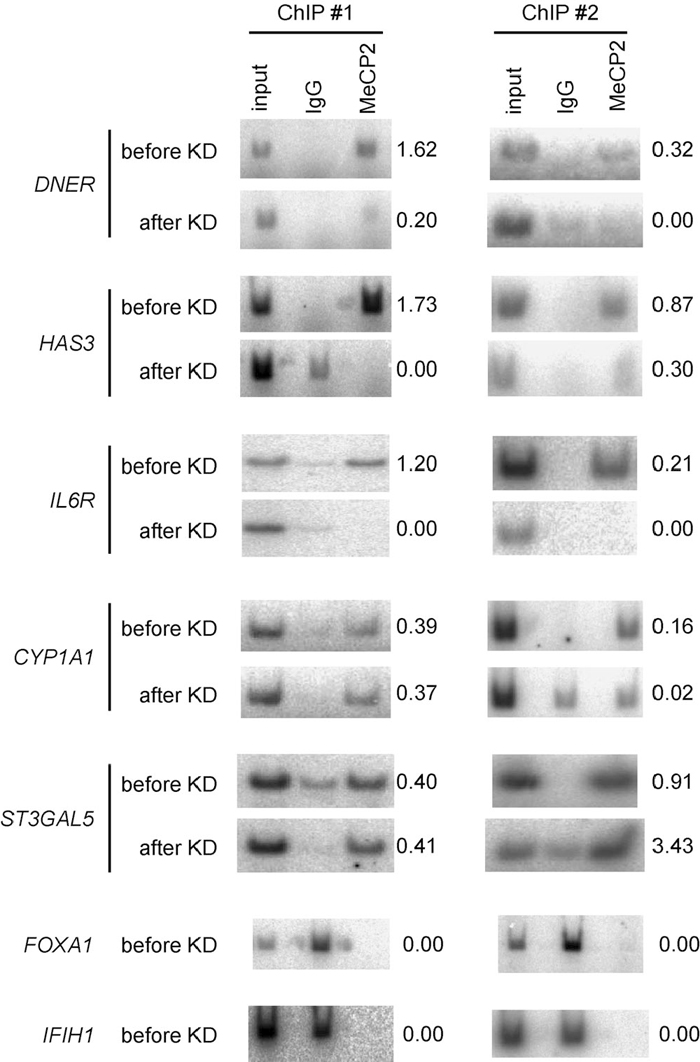 View Details | Fig. 4 Representative results of ChIP-PCR analysis. DNER, HAS3, IL6R, CYP1A1, and ST3GAL5 were bound by MeCP2 before KD, and MeCP2 binding was reduced for DNER, HAS3, and IL6R after KD. Contradictory or inconsistent results were noted for CYP1A1 and ST3GAL5 in different ChIP assays after KD. Since FOXA1 and IFIH1 were not bound by MeCP2 before KD, we did not perform ChIP assays for these genes after KD. Five independent ChIP assays were performed. We defined MeCP2 binding as positive when at least three positive bands were obtained by ChIP-PCR with a γ-32P-labeled forward primer. MeCP2 binding was defined as negative when at least three bands were absent relative to the input. The relative intensity of MeCP2 to input is shown to the right of each gel. |
Thus, among the seven selected genes, only for two genes, DNER and HAS3, were DNA methylation and constitutive MeCP2 binding before and reduced MeCP2 binding after KD all observed (Table 4). Although the CGIs of IL6R, CYP1A1, and ST3GAL5 were not methylated, MeCP2 constitutively bound to the CGIs of these genes before KD. After KD, however, only IL6R underwent an obvious reduction in MeCP2 binding. The CGIs of the two remaining genes (FOXA1, and IFIH1) were also unmethylated, and were not bound by MeCP2 at all.
 View Details | Table 4 DNA methylation and MeCP2 binding of 7 selected genes |
To characterize the genes up-regulated by MECP2 KD (putative MeCP2 target genes), we searched for gene ontology (GO) data relating to these genes. The GO Project provides a controlled vocabulary that used to describe the biology of a gene product. There are three independent sets of vocabularies, or ontologies, which describe the molecular function of a gene product, the biological process in which the gene product participates, and the cellular component where the gene product can be found. GO data comprises “terms”, each of which is assigned to one of the three ontologies. Thus, a gene product might be associated with not only one or more ontologies but also several terms.
Ontologies of biological process, cellular component, and molecular function were associated with 43, 41, and 42 of the genes upregulated by MeCP2, respectively (Table 5). The frequencies of each GO for the group of 49 up-regulated genes did not differ significantly from the frequencies for all human genes. A total of 223 terms were obtained for the 49 genes. Only the 14 terms that were associated with more than five genes are shown in Table 5. The frequency of “extracellular space” as a cellular component, including GDF15, IL1B, IL32, LOXL2, MMP1, MMP7, MMP9, OVGP1, and SULF2, was significantly higher for the up-regulated genes than for all human genes (18.4% vs. 2.7%). This suggests that MeCP2 preferentially represses a certain group of genes, the products of which exist in the extracellular space. Other terms, including “integral to plasma membrane”, “cell adhesion”, “endoplasmic reticulum”, and “nucleus”, which were related to the cellular component, and “calcium ion binding” and “extracellular region”, which were related to the molecular function, showed a tendency to be higher for the up-regulated genes than for all human genes, with p values of less than 0.05.
 View Details | Table 5 Ontology of genes up-regulated by MeCP2 knockdown |
In this study, we found that half of the 49 genes up-regulated by MECP2 KD did not have a promoter CGI (Table 3), which is believed to be important for epigenetic gene repression. Furthermore, the promoter CGIs in some genes were not always methylated or bound by MeCP2 (Table 4). A recent study using a human neuronal cell line showed that only 5.9% of MeCP2-bound sites overlapped with CGIs and that only 6.0% of MeCP2-bound promoters were highly methylated, being consistent with our results (Yasui et al., 2007). We also found that, in living cells, MeCP2 bound to the CGIs of three of the seven genes that were up-regulated by MECP2 KD, and that these CGIs were mostly unmethylated. Expression of the genes downstream of these CGIs increased after MECP2 KD, in particular expression of IL6R, for which MeCP2 binding was completely lost after KD, and a 5.3-fold increase in expression was found (Fig. 1d and Fig. 4, Table 4). This indicates that non-canonical, DNA methylation-independent gene repression is being effected by MeCP2, which differs from the DNA methylation-dependent means of canonical MeCP2 gene repression. It has been reported that MeCP2 can bind to unmethylated nucleosomal arrays and condense them in cell-free system, depending on the region(s) of the protein other than the MBD (Georgel et al., 2003). The affinity of MeCP2 for methylated DNA is only up to 3.3-fold stronger than that for unmethylated DNA (Ballestar et al., 2000; Fraga et al., 2003). Furthermore, a recent study showed that 28% of unmethylated promoter CGIs were bound by MBD proteins in cancer cell lines, some of which were bound by MeCP2 (Lopez-Serra et al., 2006). These reports support our finding that MeCP2 binds to unmethylated promoter CGIs and represses gene expression in living cells, representing a methylation-independent form of repression. Although the number of genes analyzed was not large, the data we gathered suggested that at least some of the target genes of MeCP2 are regulated in this non-canonical manner. Whether this non-canonical gene repression is associated with histone modification, such as deacetylation of H3/H4, methylation of H3K9, or other chromatin remodeling factors, remains to be clarified. It is also unclear whether other MBD proteins repress gene expression in a similar way. Interestingly, we found two genes (FOXA1 and IFIH1) in which the CGIs were not methylated and not bound by MeCP2, but for which expression was elevated by MECP2 KD. FOXA1 had three other CGIs within –5 kb to +5 kb relative to the transcription initiation site, but IFIH1 did not have any other CGI within the region. The other CGIs might be responsible for MeCP2 mediated repression of FOXA1. Or non-CGI region might be involved in repression of these genes. As another possibility, MeCP2 might function via an indirect repression mechanism.
Two of the selected genes that were up-regulated by MECP2 knockdown, DNER and HAS3, were methylated and bound by MeCP2 in LU65 cancer cells, indicating that these genes are repressed in the canonical DNA methylation-dependent manner. Since these genes were specifically methylated in LU65 cells but not in BEAS-2B bronchial epithelium cells, it seems likely that aberrantly methylated CGIs might be preferentially targeted in the canonical manner by MeCP2. Because methylation was not altered by MECP2 KD, irrespective of gene expression, we conclude that CGI methylation alone is not sufficient to repress gene expression. Heterochromatinization due to the recruitment of MBD proteins and chromatin modification factor(s) is needed for repression, as shown in our previous study (Zhao et al., 2005).
To date, there have been attempts to identify MeCP2 target genes in mice and humans (Bienvenu and Chelly, 2006). The number of target genes identified that are expressed in neurons or brain tissue, however, was too small to permit GO analysis. We performed GO analysis for the 49 putative MeCP2 target genes identified. This is the first GO analysis of MeCP2 target genes. The findings suggest that MeCP2 represses specific groups of genes, those that express products that exist in the extracellular space, in human LU65 cells. Some of them have been reported to possess anti-tumor functions; GDF15 codes an antitumorigenic and proapoptotic protein (Baek et al., 2001; Lee et al., 2005; Jang et al., 2006). Over expression of IL1B occurs cell apoptosis in MCF7 cancer cell (Roy et al., 2006). LOXL2 was markedly reduced in malignantly transformed cell lines (Rost et al., 2003). SULF2 is a potent inhibitor of myeloma tumor growth in vivo (Dai et al., 2005). The expression of certain MMPs including MMP9 provides a protective effect in cancer progression though MMPs are generally believed to stimulate tumor growth, invasion, and metastasis (Martin and Matrisian, 2007). Thus, the repression of these genes by MeCP2 could be beneficial to LU65 cancer cell. More target genes must be identified from different cell types or tissues to allow a more precise characterization of the MeCP2 target genes and to permit a better understanding of the biological roles of MeCP2.
This study was supported in part by a Grant-in-Aid for Scientific Research (C) (No. 18590313) from the Japan Society for the Promotion of Science, a Grant for Child Health and Development from the National Center for Child Health and Development, and by grants provided by the Takeda Science Foundation, the Nakayama Science Foundation, the Promotion of Science and Public Trust Surgery Research Fund, and the Ichiro Kanehara Foundation.
|