| Edited by Ali Masoudi-Nejad. Bahram Goliaei: Corresponding author. E-mail: goliaei@ibb.ut.ac.ir |
Pattern discovery or motif finding in DNA sequences is one of the most challenging problems in both molecular biology and computer science. Indeed, motifs that correspond to oligonucleotide patterns, and are significantly overrepresented in a set of functionally equivalent sequences, are good candidates for some crucial activities in regulation of gene expression (Pavesi et al., 2001). Gene expression is the process whereby a gene is transcribed to form an RNA sequence. This sequence is then used to produce the corresponding protein sequence. In order to start the gene expression process in eukaryotes, a macro molecule called transcription factor (TF) will bind to a short subsequence in the promoter region of the gene. This subsequence is called transcription factor binding site (TFBS). A transcription factor can bind to several binding sites in the promoter regions of different genes to make these genes co-regulated and expressed. The identification of TFBSs and other genomic regulatory elements that control gene expression, and characterization of their interaction with the respective TFs, are not only at the heart of understanding the network of gene interactions but also explain the origins of organismal complexity and development. The TFBSs and other genomic regulatory elements with specific structure and function are called motifs or signals. The motifs are usually recognized by the subsequences with most occurrences in a set of unaligned DNA sequences. The problem of finding them is called pattern discovery or motif finding (Pevzner, 2000).
The goal of motif finding problem is to find all subsequences, from a set of promoter regions without knowing the positions of the binding sites. It can be formulated as follows: Given a sample set of sequences {s1,...,st}, find all the subsequences of length ℓ (ℓ-mers) that occur with some mismatches in the sample set (Pevzner, 2000). There are two common models for motif representation: String (or consensus) representation and Position Frequency Matrix (PFM) (or profile) representation (Stormo, 2000). String representation uses an ℓ-mer sequence of symbols (or nucleotides) A, C, G and T to describe a motif. PFM representation contains the probability for each nucleotide at each position i.e. the value of j-th column in each row gives us the probability of occupying the j-th position of the motif by symbols A, C, G or T.
Current approaches to pattern discovery focus on monad patterns corresponding to relatively short contiguous strings that surprisingly appear (with some mismatch) many times (Bailey and Elkan, 1995; GuhaThakurta and Stormo, 2001; Hertz and Stormo, 1999; Lawrence et al., 1993; Pavesi et al., 2001; Stormo, 2000; Tompa et al., 2005). On the other hand, it is experimentally shown that the transcription regulatory mechanism in eukaryotes and especially in higher organisms is inherently much more complex than the moderation of gene expression levels by binding of one single TF to the gene promoter region (GuhaThakurta and Stormo, 2001; Yuh et al., 1998). In many cases, TFs do not work alone, therefore, regulation is resulted by the cis-regulatory action of several factors.
Many of the actual regulatory signals are groups of monad patterns that occur relatively near each other and create a composite pattern (Gelfand et al., 2000; GuhaThakurta and Stormo, 2001; van Helden et al., 2000; Klepper et al., 2008). These patterns are often associated with co-regulated genes that share two or more transcription factors (Ausiello et al., 2008).
One famous type of composite patterns is dyad signal which is a pair of monad patterns that appear within a fixed distance from each other. A possible approach for the detection of dyad patterns is to find each part (monad) of the patterns separately and reconstruct the dyad pattern. A difficulty in discovering the composite signals is that one of the components of the monad signals in the group may be too weak. This weakness makes finding the dyad patterns using the traditional monad-based approach difficult. Since the traditional monad-based motif finding algorithms usually output one high score pattern, they often fail to find dyad regulatory signals consisting of weak monad parts (Eskin and Pevzner, 2002). Some TFs like Gal4p bind to a pair of conserved tri-nucleotides that are separated by a spacer of fixed length but variable contents. The short conserved tri-nucleotides correspond to residues that enter into contact with the DNA-binding domain of the transcription factor. Their pairing is due to the fact that the transcription factor forms a dimmer, with each unit binding to a similar small element, accounting for the symmetry of the site. The fixed spacing in the DNA site is due to the existence of a linker domain in the transcription factor, separating the DNA-binding and dimerization domains. Since the factor generally does not interact with the spacer directly, this region has a much less conserved bases composition than the two contact sites. Such spaced dyad elements are common for the large class of TFs. The monad pattern finding approaches are inefficient in detecting such space pairs (GuhaThakurta and Stormo, 2001).
A better approach would be to detect both parts of a dyad pattern simultaneously. There are some algorithms that find dyad patterns simultaneously. SMILE uses a suffix tree based approach for discovering structured motifs (Marsan and Sagot, 2000). For finding dyad motifs of the length ℓ, the suffix tree is traversed by Depth First Search method. Then all the patterns corresponding to the paths from root to the nodes in the level ℓ are produced. A pair of patterns is reported as a dyad motif if part of this pair represents a valid motif. The algorithm is efficient enough to be able to infer site consensus, such as, promoter sequences or regulatory sites, from a set of unaligned sequences corresponding to the non-coding regions upstream from all genes of a genome. MITRA is another method that looks for dyad patterns using mismatch trees (Eskin and Pevzner, 2002). In this algorithm first all monad patterns are produced and then a set of virtual monad patterns (concatenation of the monad parts of the composite pattern) which represents the possible instances of the composite pattern is created. Later an exhaustive monad pattern discovery algorithm is applied to virtual monad patterns, and a graph where each vertex of it is an ℓ-mer in the sample is created. Finally the composite patterns are extracted from this graph based on pair wise similarity. MITRA is able to discover weaker dyads and composite motifs of combined length over 30, unlike its previous exhaustive pattern discovery methods. Dyad-analysis (van Helden et al., 2000) detects statistically significant motifs by counting the number of occurrences of each possible spaced pair and compares these with the expectation. Clearly, if the occurrences of pairs are more than the occurrences of random sequences, then these pairs are reported as motifs. The shortcoming of this algorithm is that there are no variations allowed within an oligonucleotide. YMF (Sinha and Tompa, 2003b) uses an exhaustive search to find motifs with the greatest z-scores. A P-value for the z-score is used to assess the significance of a motif. This P-value is the probability which a motif with that z-score or higher is found in random sequences of the same length and number. This algorithm is guaranteed to produce motifs with greatest z-score. In the SPACE algorithm (Wijaya et al., 2007), the sub motif notation is introduced and they are used for capturing the segments in the dyad motif. In this algorithm, the motif finding problem is formulated as a frequent sub motif mining problem. Its advantage is flexibilities in terms of allowing mismatches and provides an efficient method to find the pattern. All the above five methods use consensus representation for showing the motifs. In Bioprospector algorithm, zero to third-order markov background models and the Gibbs sampling strategy for finding the dyad motifs are applied (Liu et al., 2001). The significance of each found motif is judged based on a motif score distribution estimated by a Monte Carlo method. The algorithm was successful in finding motifs for binding of RAP1 protein in yeast, TATA-Box motif in Bacillus subtilis and CRP protein binding site in Escherichia coli. CoBind (GuhaThakurta and Stormo, 2001) finds dyad patterns by maximizing the joint likelihood of the occurrences of two binding sites of motifs in the process of describing the position weight matrices for the two binding motifs. In this algorithm the Gibbs sampling strategy is utilized to model the cooperatively between two transcription factors. In cases where binding site patterns are weak and can not be identified by other available methods, Co-Bind by virtue of modeling the cooperatively between factors, can identify those sites efficiently. AlignACE (Hughes et al., 2000) also uses a Gibbs sampling strategy for finding a set of motifs. In this algorithm an iterative masking procedure is used to allow multiple distinct motifs to be found within a sample dataset. AlignACE offers both efficiency and convenience. Its high signal-to-noise ratio referentially reduces false positives in the program output, while iterative masking uncovers multiple, distinct sequence motifs within a single dataset. This algorithm is built into a multi-level sequence analysis program that highlights gene specific regulatory elements for further analysis. MEME (Bailey and Elkan, 1995) uses statistical modeling techniques, E-Value, to automatically choose the best width, number of occurrences, and description of each motif. It uses an Expectation Maximization algorithm to fit a two component finite mixture module to the sequence data, the two components being the motif and the non motif parts all of sequences. The aim of MEME is to discover new motifs in a set of biopolymer sequences where little is known in advance about any motifs that may be present. All the latter four algorithms use a profile represented for showing motifs.
Broadly, motif (monad or dyad) finding algorithms can be divided into two categories: deterministic and nondeterministic (Paul and Iba, 2006). Most deterministic algorithms use exhaustive search method to specify some classes of the allowable patterns for motifs. Pratt (Jonassen et al., 1995) and TEIRESIAS (Rigoutsos and Floratos, 1998) are examples of two deterministic algorithms that use regular expression rules to identify motifs. On the other hand, most nondeterministic motif discovery algorithms are non-exhaustive and stochastic in nature. These algorithms find different motifs in different runs that may or may not be the optimal one. Some popular stochastic motif discovery tools are MEME (Bailey and Elkan, 1995), CONSENSUS (Hertz and Stormo, 1999), Gibbs sampling (Lawrence et al., 1993), MotifSampler (Thijs et al., 2001), AlignACE (Hughes et al., 2000), and BioProspector (Liu et al., 2001). Excellent history of development and application of computer algorithms for motif finding can be found in Das and Dai (2007) and Sandve and Drabløs (2006).
As mentioned, some of motif finding algorithms use consensus representation and other use profile representation. The advantages of algorithms with consensus representation is that if we have less mismatches between motifs instances and original motifs, then these algorithms guarantee to find the best scoring pattern. In other hand, consensus patterns in general can not distinguish between conserved and non conserved positions, and many motifs in real biological data can not be adequately described in this way. For example the motif with multiple bases in one position, can not be shown by consensus representation without increasing the alphabet including IUPAC symbols. Even using the IUPAC symbols, the patterns can not represent different degree conservation among different bases. Definitely, the algorithms using these representations are disable to find such motifs. In opposite, profiles are very powerful representation of the motifs and the scoring function is motivated by an underlying probabilistic model. But with less number of sequences and scattered mismatches, this method of representation is not suitable. A major disadvantage is that the problem of discovery the best profile is inherently very difficult. Most algorithms which use this representation can be characterized as performing stochastic optimization or greedy searches and give no grantees to find the best profile or even close to the best profile.
Recently, another stochastic approach, genetic algorithm, has been used for identification of motifs in the multiple unaligned DNA sequences (Che et al., 2005; Fogel et al., 2004; Gertz et al., 2007; Liu et al., 2004; Paul and Iba, 2006; Stine et al., 2003; Wei and Jensen, 2006). In these evolutionary computation methods, different models of fitness calculations are used and only one motif per sequence is assumed. However, multiple similar motifs may be existed in a sequence, and the identification of those motifs is also important for the identification of a single motif per sequence. Also, none of these genetic algorithms can be used for predicting the dyad motifs.
In this paper, we propose a novel algorithm, GA-DPAF (Genetic Algorithm for Dyad PAttern Finding), as a solution to the dyad pattern finding problem on the basis of the genetic algorithm. All the details of this algorithm are discussed on dyad motifs but it can be easily extended for composite motifs. Our genetic algorithm employs a multi-objective fitness function, and two points crossover and mutation operations. The individuals required for the population pool used in the genetic algorithm are optimized by Gibbs sampling method. The main advantages of our proposed method are the handling of the dyad motifs and the ability of identifying multiple positions of the motif instances of an original motif. Also, the algorithm uses both consensus and profile motif representation, therefore this algorithm supports the advantages of each representation together. We ran the program based on the real datasets and compared the obtained results with AlignACE (Hughes et al., 2000), MEME (Bailey and Elkan, 1995), YMF (Sinha and Tompa, 2003b), and MITRA (Eskin and Pevzner, 2002) to demonstrate the effectiveness of our proposed method. Note that the main reason for choosing these tools for comparison is due to their availability and the accuracy of their results comparing with the other existing tools.
In this section, we introduce some definitions and notations that are used in this paper. Here, a sequence is a string on a given alphabet Σ thus Σ = {A, C, G, T} for DNA, and a substring s[j]...s[j + ℓ – 1] with the length ℓ of a given string s is referred as a subsequence of length ℓ (or ℓ-mer). Let S = {s1,...,st} be t sequences of length n, we desire to find common subsequences or unknown motifs in S. As mentioned there are two representation models for motifs, consensus and profile, here, these two models are employed for the motif representation.
We use the term motif instance to mean a subsequence of the length ℓ in each sequence of the set S with mutations at some positions of an original motif. Now, if the original motif is represented by a sequence e, for finding motif instance in each sequence si of S, a subsequence of length ℓ is selected based on the most matches with the sequence e. The number of matches between two sequences x and y with the length ℓ is defined as: 
where 
For example, for the given original motif e = AGATT with length ℓ = 5, if we have the following sequences
s1 = gggagtcccgtAGCTTaggcctcgg
s2 = aACAATaccaacgcaactctagggc
s3 = aaaccccaaACATAaaacgccgcta
s4 = cttaccatcGCATAcgaggacagaa
then the motif instance of e in s1 with 1 mismatch is AGCTT, respectively, in s2 with 2 mismatches is ACAAT, in s3 with 2 mismatches is ACATA, and in s4 with 3 mismatches is GCATA.
If the original motif is represented by position frequency matrix W, for finding a motif instance in each sequence si of S, a subsequence with the length ℓ of each sequence from the set S is selected based on the maximum score with the W matrix. The score between a subsequence x of length ℓ and the W matrix is defined as follows: 
where W[α,i] is equal to the occurrence probability of character α ∈ Σ in i-th position of the original motif, and b[α] is the occurrence probability of character α in the set S. For example, the PFM corresponding to the above motif instance is given in below. 
The set U ={u1,...,ut} is called motif instance set where each ui is a motif instance in si of the original motif. The set P = {p1,...,pt} is called motif instance position set where pi is the start position of the motif instance ui in si. The pattern shared by the subsequences ui (1 ≤ i ≤ t) of the motif instance set U = {u1,...,ut} is dubbed a consensus or a consensus motif and shown by δ. Obviously, for the sequences and motif instances of the mentioned example the motif instance set is equal to U = {u1,u2,u3,u4,} where u1 = AGCTT, u2 = ACAAT, u3 = ACATA, and u4 = GCATA and the motif instance position set is equal to P = {12,2,10,10}. Respectively, their corresponding consensus is equal to δ = ACATA.
Note that in motif finding problem the original motif is unknown and we desire to find it. For this reason, in the given sequence set S, subsequences with few mismatches are selected as a motif instance set. The selection criteria can be different in various motif finding algorithms. Eventually the consensus motif of the selected motif instance set is considered as an original motif.
Similar to the above definitions, dyad motif, dyad motif instance, dyad motif instance set, dyad motif instance position set, and dyad consensus motif are defined. For a dyad motif, we are interested in discovery of two monad patterns that occur in a certain length apart. We use the notations e* = ea + eb, to denote a dyad motif e* of length ℓ, which consists of two monad patterns ea and eb of length ℓa and ℓb by a distance d, dmin ≤ d ≤ dmax, for predefined bounds dmin and dmax. A dyad motif instance set of the motif e* = ea + eb is a set of sequences that can be denoted by U* = Ua + Ub = {u1a + u1b,...,uta + utb} = {u1*,...,ut*} with the length ℓ = ℓa + ℓb, and is constructed by concatenation of the two motif instances Ua = {u1a,...,uta} and Ub = {u1b,...,utb} of the length ℓa and ℓb respectively, such that uia and uib are selected from the sequence si by a distance d, (dmin ≤ d ≤ dmax). The set P* = {(p1*, d1),...,(pt*, dt)} is called the dyad motif instance position set where pi* is the start position of the motif instance uia (ui* = uia + uib) in si and di is the distance between the two motif instances uia and uib. In this notation the instance motif ui* = uia + uib with the starting position pi* of uia in the sequence si = si[1]...si[n], is shown as the subsequence si[pi*]...si[pi* + ℓa –1]si[pi* + ℓa + di –1]...si[pi* + ℓa + ℓb + di –1]. Also, if di = 0 (1 ≤ i ≤ t) then the motif instance is monad and therefore P* = P and U* = U. For example, for the given original dyad motif e* = AGAATC of length ℓ = 6, where ea = AGA of length ℓa = 3 and eb = ATC of length ℓb = 3, if we have the following sequences
s1 = ttttcaattcAGCattAGCattgtac
s2 = agactgtCGAcATCcacatcccctat
s3 = tcttaaaCGAcaATAttttacacaca
s4 = cacccgtagGGAggAGCaggaaagtc
then the dyad motif instance set is equal to U* = {u1*, u2*, u3*, u4*} where u1* = u1a + u1b = AGCAGC, u2* = u2a + u2b = CGAATC, u3* = u3a + u3b = CGAATA, u4* = u4a + u4b = GGAAGC and the dyad motif instance position set is equal to P* = {(11,3),(8,1),(8,2),(10,2)}. Respectively, their corresponding consensus is equal to δ = CGAAGC.
Genetic Algorithm (GA) is an adaptive method which may be used to solve search and optimization problems (Goldberg, 1989). It is based on the genetic processes of biological organisms. Over many generations, natural populations evolve according to the principles of natural selection and survival of the fitness, first clearly stated by Charles Darwin. Genetic algorithm works with a population of the individuals, each representing a possible solution to a given problem. Fitness value is assigned to each individual according to how good is the corresponding solution for the problem. The highly fit individuals are given opportunities to reproduce, by crossing with other individuals in the population. This produces new individuals as offsprings that share some features taken from each parent. The least fit members of the population are less likely to be selected for reproduction, and so die out after a while. A whole new population of possible solutions is thus produced by selecting the best individuals from the current generation, and having them mate each other to reproduce a new set of individuals. This new generation contains a higher proportion of the characteristics possessed by the good individuals of the previous generation. If the GA has been designed well, it can be proved that, the population will converge to the optimal solution of the given problem (Goldberg, 1989).
The steps of our genetic algorithm, GA-DPAF, are illustrated in Algorithm 1. The definitions of population, the fitness function, crossover and mutation operators are given below. The input parameters of algorithm are the set S = {s1,...,st} (in which motifs should be found), n (the length of each sequence si), ℓa and ℓb (the length of the first and the second parts of the dyad motif respectively), and dmin and dmax (minimum and maximum distances between motifs in each sequence).
 View Details | Algorithm 1 Genetic algorithm GA-DPAF. |
In our genetic algorithm, each individual is a dyad motif instance position set P*, which is created in each step of the algorithm (randomly in initial step and by crossover and mutation in other steps) and is improved by the Gibbs sampling method. For this process first a dyad motif instance set U* is made from P* and then we randomly pick i-th sequence, and discard i-th dyad motif instance from U* and create PFM W based on the other t–1 dyad motif instances in U*. Now for each j (1 ≤ j ≤ |si|–ℓa –ℓb – d) and d (dmin ≤ d ≤ dmax), we compute score(s',W) where s' = si[j]...si[j + ℓa–1]si[j + ℓa + d–1]...si[j + ℓa + ℓb + d –1]. The values of j and d corresponding to the s' with the highest score, score(s',W), are selected and are replaced with (pi*,di) in the set P*. This process is repeated until fitness of U* (the fitness function is described in the next subsection) can not be improved. The final dyad motif instance position set P* is considered as an original individual.
Initial population of our algorithm consists of 
number of individuals that each individual is a dyad motif instance position set. Initial population is made based on the algorithm shown in Algorithm 2. In this algorithm Steps 9, 10, and 11 are performed in the similar method mentioned in the Definitions and Notations section, and Step 12 is performed with Gibbs sampling method discussed in above.
 View Details | Algorithm 2 Initial population algorithm. |
Each individual is evaluated depending on the feature of the problem. We propose three different partial fitness functions for the motif discovery problem, and the final fitness function is computed by the summation of these partial fitness functions: 
where FSP is the sum of pairs, FIC is a function defined based on the information content (IC) of the motif instance set, and FNM is the number of matches between the motif instance set and its corresponding consensus motif. Note that, although these partial fitness functions have overlaps but the obtained results showed that using the sum of them performs better than each in single.
We extract a dyad motif instance set Ug* = {uga,1 + ugb,1,...,uga,t + ugb,t} = {ug*,1,...,ug*,t} from the sequence set S, for any existing individual Pg* = {pg*,1,dg,1),...,(pg*,t,dg,t)} 1 ≤ g ≤ m in the pool and then we compute each fitness function as follows.
FSP: The number of mutual matches between the sequences in each motif instance set is computed by this function. In other words, for any Ug* = {ug*,1,...,ug*,t} the numbers of mutual matches between ug*,1,...,ug*,t are computed. Therefore, the fitness function FSP for the motif instance set Ug* = {ug*,1,...,ug*,t}, is equal to: 
where ug*,i and ug*,k are the i-th and k-th dyad motif instances of the motif instance set Ug* respectively, and M(ug*,i,ug*,k) is the number of matches between ug*,i and ug*,k.
FIC: For the motif instance set Ug* = {ug*,1,...,ug*,t}, the corresponding position frequency matrix W is constructed. Later the fitness function FIC is computed as follows: 
FNM: This function evaluates the number of matches between a given motif instance set Ug* and its corresponding consensus motif δg. The number of matches between this consensus motif δg and the motif instance set Ug* is evaluated as: 
where ug*,i is the i-th dyad motif instance of the motif instance set Ug*.
To consider these three different functions as a fitness function, we first scale them in the range of [0,1] and then totally add them up. For scaling, we consider the following points:
1. The maximum value of FSP is equal to 
Therefore, we consider 
for scaling in the range of [0,1].
2. The maximum value of FIC is equal to MaxIC = 2 × ℓ.
Therefore, we consider 
for scaling in the range of [0,1].
3. The maximum value FNM is equal to MaxNM = t × ℓ.
Therefore, we consider 
for scaling in the range of [0,1].
Our genetic algorithm uses a two-point crossover with the rate pc. For this reason, first we select two individuals Pg* and Ph* from the population pool by roulette wheel selection and consider them as candidate parents. Then, these two parents are crossed over based on the Crossover procedure shown in Algorithm 3. Mutation operation is done with the rate pm on the best individuals. This operation on an individual P* is shown in Algorithm 4. As we can see the mutation operation is done based on the information content (IC) of position frequency matrix corresponding to the individual P*.
 View Details | Algorithm 3 Crossover algorithm. |
 View Details | Algorithm 4 Mutation algorithm. |
The GA-DPAF algorithm terminates on two conditions: after N iterations or when few consecutive generations yield similar best individuals. After the termination of the
algorithm, k top individuals 
with the best fitness among all the individuals are selected as final dyad motif instance position sets. Let P1*...Pk* be these best individuals, and then position frequency matrix Wi (1 ≤ i ≤ k) is created for each of these individuals. Later score(x,Wi) is calculated for each subsequence x with the length ℓ from the set S. The calculated score is normalized as follows: 
Where 
Now, between all the subsequences of the set S, the sequence x with the best normalized score is considered as an original motif, if the normalized score x and the corresponding PFM W (NScore(x,W) becomes larger or equal to a cutoff. In the next section, we show that for strong motifs this cutoff is 0.9 and for weak motifs this cutoff is 0.8. By these values, we can obtain some dyad motifs in each sequence.
Evidently, the given genetic algorithm can be easily modified for obtaining composite patterns with more than two, and only the crossover and mutation operations should be extended.
Here, we discuss the time and space complexity of our algorithm in each iteration, for finding dyad motif of length ℓ in t given sequences of length n, such that maximum valid distance between the two parts of the dyad motifs is d. With respect to the Algorithm 2, generation of the initial population takes O(t2ℓdn). The crossover operation shown in Algorithm 3, takes O(tℓdn), and respectively the mutation operation demonstrated in Algorithm 4 is performed in O(tℓdn) time complexity. Since the algorithm GA-DPAF given in Algorithm 1 uses crossover and mutation in each step, thus the time complexity of each iteration is O(t2ℓ2dn). Because of the evolutionary feature of our algorithm, the number of iteration in our algorithm depends on the applied dataset.
In other hand, for storing the t sequences of length n we need O(tn) space complexity. Also, the population used in our algorithm represents both consensus motifs and position sets, and the storage complexity of these two types are O(ℓm) and O(tm) (m is the population size) respectively. Thus the total space complexity of GA-DPAF is equal O(tm + tn + ℓm).
The algorithm GA-DPAF is implemented in Perl with compiler version 2.3 and is developed on a PC with four dual-core processor AMD 2.0 MHz running Ubuntu 8.04. The code is portable and can be compiled and run under the UNIX and Windows environment. The source of program is available to anyone upon the Email request from the corresponding author.
In this section, we intend to compare our algorithm to other motif finding algorithms on different real datasets. The comparison is performed in nucleotide levels regarding their positions in the main sequences. For this reason, we first introduce the following criteria for the comparison (Tompa et al., 2005).
1. nTP is the number of nucleotide positions in both known sites and the predicted sites.
2. nFP is the number of nucleotide positions not in the known sites but in the predicted sites.
3. nFN is the number of nucleotide positions in known sites but not in the predicted sites.
Regarding the above criteria and recall from Pevzner and Sze (2000), measurement Nucleotide Performance Coefficient (nPC) for the evaluation of the algorithms is defined in the nucleotide level of the predicted sites and is equal to 
As we can see, nPC ≤ 1 and the higher value of nPC shows that the known sites and the predicted sites are more similar. Obviously, if the predicted sites were equal to the known sites then nPC is equal to one. We need a way for summarizing the performance of a given motif finding program over all datasets. Recall from Tompa et al. (2005), for each program, the measurement nPC over all datasets is obtained and the performance of each program on all datasets are compared by the following methods.
1. Average: Initially, the nPC value is calculated on each dataset for each program, and then the usual arithmetic mean of nPC is evaluated for each program.
2. Normalized: For each dataset, the nPC value is normalized by subtracting the mean and dividing by the standard deviation over all the programs on that dataset, and the average of these normalized scores over all datasets is obtained. This method which is also called z-score puts easy and hard datasets on the same scale.
As previously mentioned, we use a cutoff value in the final process of motif finding. This value is evaluated as follows: first some datasets are considered as the training set, and our algorithm is performed on these datasets. Clearly some subsequences are reported as potential motifs. The position frequency matrixes corresponding to these potential motifs are computed. Later each sequence x ∈ S of length l is aligned by the PFMs, and their corresponding scores are evaluated by the function NScore(x,W). The initial cutoff is set to 1 and the sequences with the score less than this cutoff are dropped, the others are regarded as new potential motifs. With respect to the actual TFBS of the training set, the value of nPC is computed. Then the value of cutoff is decreased by the rate of 0.02. Performing the above process for the newly potential motifs, we obtain a new nPC value. If the new value of nPC was more that the old one the whole process is repeated for smaller value of cutoff, until to get a cutoff which decreases the values of nPC. The previous cutoff before the last decrease is selected as cutoff value. As a consequence of the above experiment, we observed that for strong motifs 0.9 is a suitable cutoff value and for the weak motifs 0.8 is suitable. Hence, we have used the above values as our cutoff in our tests.
In the next sub-section, algorithm GA-DPAF is compared with four known programs AlignACE (Hughes et al., 2000), MEME (Bailey and Elkan, 1995), MITRA (Eskin and Pevzner, 2002) and YMF (Sinha and Tompa, 2003b) with regard to nPC. As mentioned, the reason for choosing these tools for comparison is due to their availability and the accuracy of their results comparing with the other existing tools. To investigate the ability of GA-DPAF in finding motifs from the real biological samples we tested it for finding monad and dyad motifs in different datasets. In this section, first we present the obtained result by GA-DPAF on monad motifs and compare these results with results obtained by AlignACE, MEME and YMF. Finally, we present the results obtained by GA-DPAF on dyad motifs and compare these results with the ones obtained by MITRA, AlignACE, MEME and YMF.
To evaluate the GA-DPAF algorithm on the real monad motifs, we applied it to upstream regions of orthologous genes of yeast with known motifs from the database SCPD (Zhu and Zhang, 1999). We consider the datasets from this database which is given in Sinha and Tompa (2003a). A region of gene regulation with the length 850 bp from position –800 to +50 is extracted for each regulator in this dataset. Since most of these algorithms consider 3 patterns with top score as motifs, thus we also consider 3 patterns with top scores, reported by GA-DPAF, as motifs. Therefore, comparison of our algorithm with these algorithms is performed based on these patterns.
In Table 1, the obtained nPC values for different datasets are shown (the nPC value for the other algorithms are extracted from Sinha and Tompa (2003a)). As we can see in table, the first column shows the name of regulon and the second column shows their corresponding consensus with IUPAC symbols. In this table '–' symbol is used to indicate that no consensus is reported in SCPD database. The third column shows the number of sequences and the other columns denote the nPC values for the mentioned algorithms. As we can see, our algorithm showed a higher score than the other three algorithms in most cases.
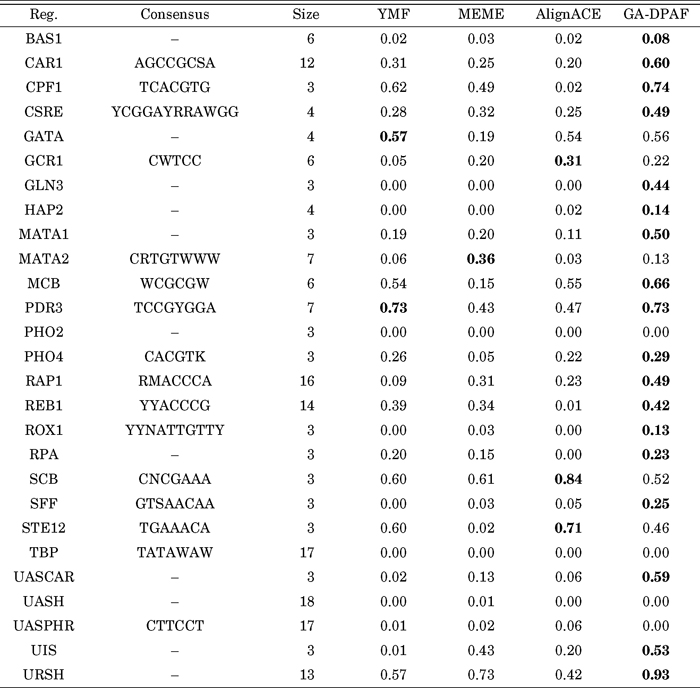 View Details | Table 1 The nPC value evaluated for YMF, MEME, AlignACE, and GA-DPAF for finding monad motifs |
For the above datasets, the Average and Normalized (z-score) values of nPC measure given in Table 1 are calculated. These values are shown in Fig. 1. Again as we can see in this figure, the GA-DPAF gives better results.
 View Details | Fig. 1 The nPC measure evaluated for the results obtained from the four algorithms on SCPD for finding monad motifs. |
GA-DPAF is tested on the real datasets that have been previously tested by MITRA, YMF, AlignACE, and MEME (Bailey and Elkan, 1995; Sinha and Tompa, 2003a). The obtained results are compared by these four algorithms. The first biological sample is formed the upstream regions involved in purine metabolism from three Pyrococcus genome studied by Gelfand et al. (2000). The signal of this data is a palindrome dyad motif consisting of two monad patterns that occurs at a distance varying from 22 to 23 nucleotides. MITRA detects this dyad motif exactly and YMF and AlignACE can not find it. Therefore, we compared the obtained results of MEME with GA-DPAF. These results are shown in Fig. 2.
 View Details | Fig. 2 The nPC measure of MEME and GA-DPAF algorithms on Purine. |
The second biological sample is composite motif URS1-UASH of yeast. GuhaThakurta and Storm (2001), described a set of yeast S.cerevisiae genes that are regulated by two transcription factors. The transcription factor binding sites corresponding to these factors occur relatively close to each other. One of the famous TFBS in this dataset is URS1-UASH. In the corresponding TFBS, although URS1 is strongly conserved and easily found with traditional monad-based approaches, UASH is too weak to be discovered with these approaches. There are 11 genes which are regulated by both URS1 and UASH transcription factor. For 10 of these genes, the two binding sites are located within the upstream regions –300 to –1. In the five of these genes, the distance between binding sites is long and in the five of genes, the binding sites are within 50 bases of each other. In Eskin and Pevzner (2002), MITRA is tested on 5 genes with close binding sites. The AlignACE, MEME, and YMF could not find UASH signal, and therefore we compared the result of GA-DPAF with the result obtained by MITRA based on nPC measure on URS1-UASH prediction. Since the results on URS1 were the same in both algorithms, therefore we just compare the obtained result on UASH as it is shown in Fig. 3.
 View Details | Fig. 3 The nPC measure of MITRA and GA-DPAF algorithms on UASH. |
The third biological sample is dyad motifs ABF1, GAL4, HAP1, HSE, MCM1, and MIG1 of yeast. In Sinha and Tompa (2003a), the algorithms YMF, AlignACE, MEME are tested on these motifs. Since most of these algorithms consider 3 patterns with top score as motifs, therefore we also considered 3 patterns with top score as the motifs using the GA-DPAF algorithm. We ran GA-DPAF on these datasets and the obtained results were compared with the results of these algorithms. The comparison results on nPC measure are shown in Table 2 (The nPC value for the other algorithms is extracted from Sinha and Tompa (2003a)). For the above regulons, the Average and Normalized of nPC values of Table 2 are calculated and shown in Fig. 4. As we can see in this figure, GA-DPAF shows better results.
 View Details | Table 2 The evaluated nPC value for YMF, MEME, AlignACE, and GA-DPAF on dyad datasets |
 View Details | Fig. 4 The nPC measure of four algorithms for finding dyad motifs. |
Consequently, by considering all the above results, and comparing GA-DPAF to the other algorithms, it is evident that GA-DPAF was more efficient in most cases, and its failure cases were just few. In brief we discuses the reason of these failures. Because of the similarity of few TFBS to background sequences, TFBS are considered as random sequences by our algorithm. For example, in the promoter region of yeast datasets, the number of occurrence of the nucleotides A and T are very frequent, therefore the TFBS with frequent occurrence number of A and T can not be detected by our algorithm.
An evolutionary method is presented for finding the dyad motifs in a given set of sequences based on the genetic algorithm. The algorithm uses a multi-objective fitness function with new crossover and mutation operations. The method is implemented and tested for finding monad and dyad motifs on the real biological datasets. The results are compared with four well-known dyad motif finding algorithms AlignACE, MEME, YMF, and MITRA.
The algorithm was successful in finding monad and dyad motifs for binding of transcription factors in Saccharomyces cerevisiae and purine metabolism motif in Pyrococcusth. Our algorithm finds dyad pattern by maximizing the fitness function which is based on information content, sum of pairs and the number of matches. Generally, there is no limitation on length of motifs for our algorithm. The effectiveness of our method is shown by the improvement of our obtained results in comparison with the other methods. In order to compare our algorithm with other known algorithms, the nPC measurement is used, and two average and normalized methods are applied for comparison. In the average method, the average of nPC values for each algorithm is computed and compared. In the normalized method, the efficiency of each algorithm on each sample is compared to the other algorithms. By the definition of nPC value, high value of nPC shows the high number of TP and low number of FN and FP. Actually, the algorithm with higher nPC value shows a better performance. In our experimental tests, we observed that for both dyad and monad motifs, the result of our algorithm have higher nPC values comparing to the others, and our method improves the sensitivity and specificity. Therefore GA-DPAF can be considered as a standard tool for monad and dyad motif finding.
The authors would like to thank the anonymous referees for their helpful and valuable suggestions.
|