| Edited by Fujio Kawamura. Kouji Matsumoto: Corresponding author. E-mail: koumatsu@mail.saitama-u.ac.jp. Satoshi Matsuoka and Michihiro Hashimoto: These authors contributed equally to this work |
Lipoteichoic acid (LTA) is a polyanionic component of the cell envelope of Gram-positive bacteria (Neuhaus and Baddiley, 2003). It is synthesized by polymerizing the sn-glycerol-1-phosphate (G1P) moiety of phosphatidylglycerol (PG) onto a diglucosyldiacylglycerol (DGDG) membrane anchor (Fig. 1). We first became interested in the LTA synthetic pathway when we found that Bacillus subtilis pgsA is an essential gene. pgsA encodes a phosphatidylglycerophosphate synthase that is a key enzyme for the synthesis of PG. It could not be disrupted, and its placement under an isopropyl-β-D-thiogalactoside (IPTG)-inducible Pspac promoter resulted in bacteria whose growth was dependent on the inducer (Kobayashi et al., 2003; Hashimoto et al., 2009). This stands in contrast to the dispensability of the pgsA gene in Escherichia coli (Matsumoto, 2001). Since in B. subtilis the pgsA gene is required for the synthesis of PG, a substrate for LTA, it would seem natural to ascribe its essentiality to its role in LTA synthesis. Gründling and Schneewind (2007) have identified the ltaS gene encoding LTA synthase in Staphylococcus aureus. Growth of an S. aureus Pspac-ltaS strain was dependent on IPTG for LTA synthesis and growth, indicating that LTA is essential at 37°C. Oku et al. (2009) have shown that S. aureus ltaS-null mutations result in severe thermosensitivity: the mutants grow very poorly even at 30°C. Because B. subtilis has four homologues of ltaS (Gründling and Schneewind, 2007), it was not surprising that none of the homologues were among the essential genes identified by Kobayashi et al. (2003), yet it seemed possible that defective mutations of the homologues might be synthetically lethal. However, Schirner et al. (2009) have reported that a quadruple mutant defective in all the homologues could be constructed, although it grew slowly and had a cell division and separation defect. Recently, Wörmann et al. (2011) have shown that LTA is absent in the quadruple mutant. Therefore, the essentiality of the B. subtilis pgsA gene cannot be ascribed to its indispensability for LTA synthesis.
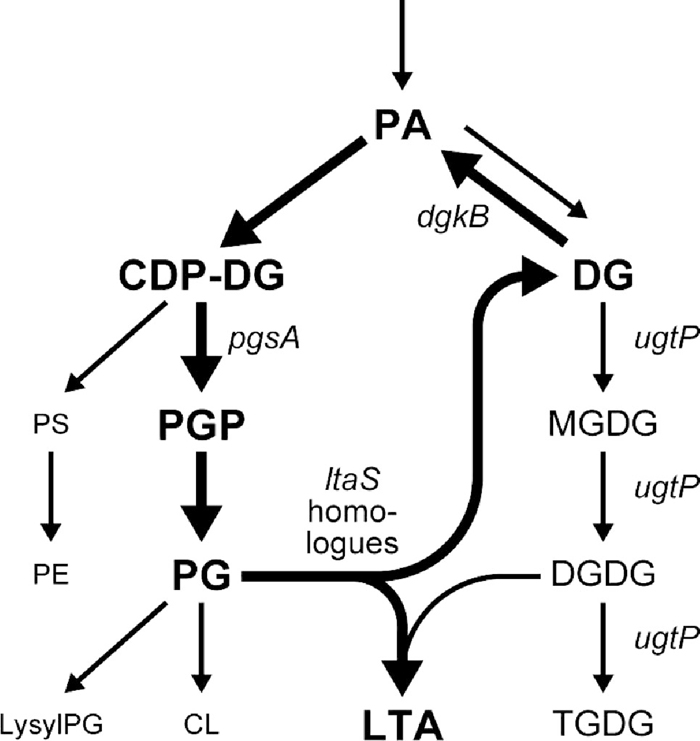 View Details | Fig. 1 Synthetic pathways for phospholipids, glucolipids and LTA in B. subtilis. The diacylglycerol cycle (Jerga et al., 2007) for LTA synthesis is depicted with thick arrows. The genes encoding the enzymes relevant to the present study are indicated. The genes for PA and PGP phosphatase have not yet been identified. PA, phosphatidic acid; PS, phosphatidylserine; PE, phosphatidylethanolamine; PGP, phosphatidylglycerophosphate; PG, phosphatidylglycerol; lysylPG, lysylphosphatidylglycerol; CL, cardiolipin; DG, diacylglycerol; MGDG, monoglucosyldiacylglycerol; DGDG, diglucosyldiacylglycerol; TGDG, triglucosyldiacylglycerol; LTA, lipoteichoic acid. |
The synthesis of LTA releases diacylglycerol (DG) moieties of PG. Since LTA is a long polymer, synthesis of a single LTA molecule results in the production of dozens of DG molecules. DG is converted to phosphatidic acid (PA) by DG kinase and reused in the synthesis of phospholipids, among them including PG (Fig. 1). Thus, LTA is synthesized by a cyclic reaction in which DG kinase plays a key role. The dgkB gene encoding DG kinase is essential in B. subtilis (Kobayashi et al., 2003), and repression of Pspac-dgkB leads to lethality. Jerga et al. (2007) have reported cessation of LTA synthesis and accumulation of DG in the dgkB-repressed cells. Since LTA is not essential in B. subtilis, it is unlikely that the defect in LTA synthesis is responsible for the death of the repressed cells, and the accumulation of DG is rather more likely the reason. In this study, we show that disruption of either or both of two LTA synthesis genes, yflE and yfnI, suppresses the lethality of the dgkB-repression. Accumulation of DG after repression of dgkB is suppressed by the disruption of yfnI but not yflE, suggesting that the DG accumulation is involved in, but not the immediate and sole cause of, the non-viability of B. subtilis lacking DgkB.
B. subtilis 168 derivative strains and plasmids used in this study are listed in Table 1. Deletion mutants of yflE and yfnI were constructed by replacement with spectinomycin and tetracycline resistant cassettes, respectively. To construct these mutants, yflE and yfnI, including flanking sequences extending about 800 and 1100 bp upstream, and 900 and 1100 bp downstream of the genes, were amplified by PCR using appropriate primer sets. The amplified PCR fragments were digested with appropriate restriction enzymes (for yflE, ApaI, BamHI, and PstI; for yfnI, ApaI, BamHI, and ClaI), then cloned into pBluescript II SK(+). The resulting plasmids (for yflE and yfnI) were digested with PstI and ClaI, then spc and tet genes cut out from pDG1726 and pDG1513, respectively, were ligated to produce pMH05, in which the entire coding region of yflE was replaced with spc, and pMT11, in which the region 270 bp downstream from the start codon and 170 bp upstream from the stop codon of yfnI was deleted and replaced with tet. These plasmids were used for the construction of strains MBS10 (yflE::spc) and MBS11 (yfnI::tet). Disruption of dgkB was by integration of a pMUTIN-derived plasmid (pMUTINCC) containing an internal fragment of the gene (Vagner et al., 1998) and the disrupted allele was denoted as dgkB::pMUTINCC (erm). Placement of dgkB under Pspac was by integration of a pMUTIN3 derivative containing the ribosome-binding site (RBS) and the 5′ end of the gene and this was denoted as P-dgkB (YERQp). Xylose-inducible Pxyl promoter-controlled genes with or without the 5´-terminally fused green fluorescent protein (GFP)-coding sequence were constructed on pSG1729 (Lewis and Marston, 1999) or pX (Kim et al., 1996) and integrated into the chromosomal amyE locus by selecting for resistance to spectinomycin or chloramphenicol, respectively. The genes without GFP-fusion were designed to be expressed using the RBS on the vector. The allele ugtP::kan in which ugtP was replaced with a kan cassette was from strain KP261 (Price et al., 1997).
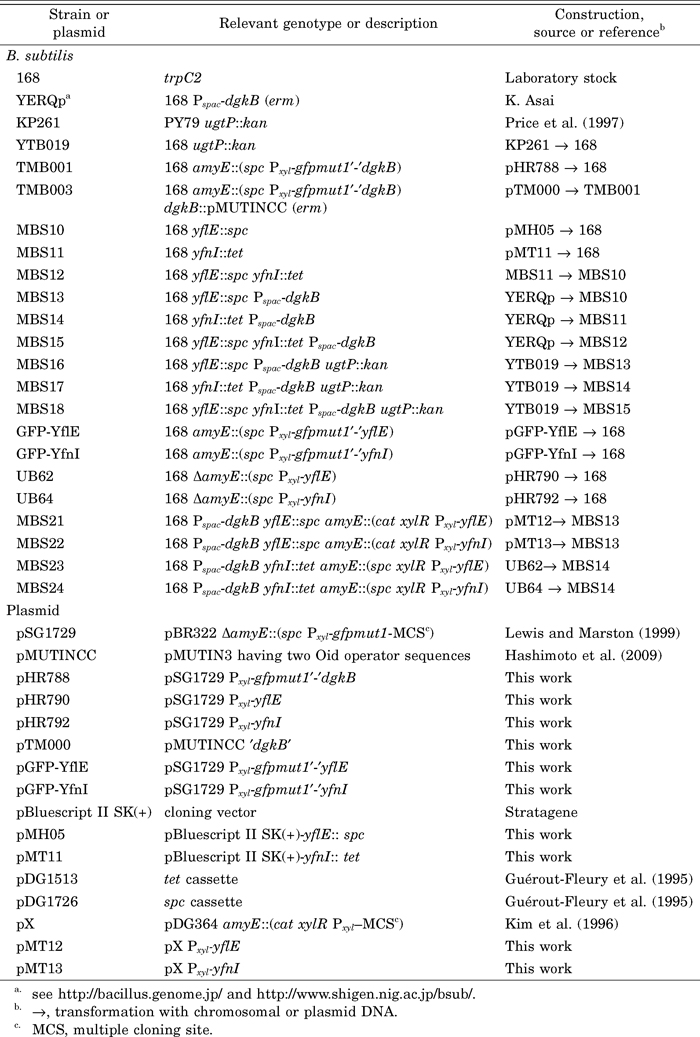 View Details | Table 1 B. subtilis strains and plasmids used in this study |
Luria-Bertani (LB) medium and buffered LB medium (Shiba et al., 2004) were used. For plates LB medium was solidified with 1.5% agar. When appropriate, antibiotics were included at the following concentrations (in μg/ml): erythromycin, 0.5; tetracycline, 10 or 15; spectinomycin, 50 or 100; chloramphenicol, 5; kanamycin, 5. Inducer IPTG was used at 1 mM, and xylose at 0.05% or 1%. Cell growth was monitored with a Klett-Summerson colorimeter (with no. 54 filter) or Shimadzu Bio Spec-mini (at OD 600 nm).
These were based on standard methods. Competent cells for transformation were prepared by one-step (Wilson and Bott, 1968) or two-step (Anagnostopoulos and Spizizen, 1961) methods. PCR primers used in this study are listed in Table 2.
 View Details | Table 2 PCR primers used in this study |
Cells were labeled with 3.75 × 104 Bq/ml of [1-14C] acetic acid (Amersham; 2.12 × 109 Bq/mmol) in LB medium at 37°C, continuously from initial inoculation for precultures to cell harvesting. Mid-exponential phase cells grown in the presence of 1 mM IPTG were washed with LB medium and diluted into LB medium with and without 1 mM IPTG to 5 Klett units. After 3 h cultivation cells were harvested, and lipids were extracted by a modified Bligh and Dyer’s method (Ames, 1968). In order to assay DG accumulation, the extracts were subjected to TLC using TLC Silica Gel 60 plates (Merck) developed with hexane/diethylether/acetic acid (50:50:1, v/v/v). Mobilities of DG and fatty acids (FA) were confirmed in preliminary experiments in which nonradioactive samples were analyzed with authentic DG (1, 2-dioleoyl-sn-glycerol; Avanti Polar Lipids) and FA (oleic acid; Wako) as standards and stained with primuline. Phospholipids and glucolipids were analyzed by two-dimensional TLC as described previously (Hashimoto et al., 2009). Lipid bands/spots were detected and quantified using an FLA-7000 image analyzer (Fujifilm).
The chromosomal DNA of the strain TMB003 [amyE::(spc Pxyl-gfp-dgkB) dgkB::pMUTINCC (erm)] was used to transform strains disrupted for either or both of yflE and yfnI, designated as MBS10 (yflE::spc), MBS11 (yfnI::tet), and MBS12 (yflE::spc yfnI::tet), selecting for erythromycin resistance of the dgkB:: pMUTINCC(erm) allele. The transformation efficiencies were compared with the efficiencies at which the erythromycin resistance of Pspac-dgkB (erm) allele from DNA of strain YERQp was introduced into the same competent cell preparations in the presence of IPTG. The transformation efficiencies of the Pspac-dgkB (erm) allele were 1.8 × 104 – 9.7 × 105/μg DNA.
Subcellular localization of GFP-fused proteins was observed under a fluorescence microscope as described previously (Nishibori et al., 2005). GFP fusions were confirmed by Western immunoblotting using anti-GFP mouse monoclonal antibody (Santa Cruz Biotechnology), horseradish peroxidase-conjugated anti-mouse IgG secondary antibody (Novagen) and ECL Advance Kit (Amersham).
In this study we focused on the yflE and yfnI genes, whose products show higher homologies to S. aureus LtaS among the four homologues (Schirner et al., 2009). Gründling and Schneewind (2007) have reported that, when expressed from a chromosomally integrated copy in ltaS-repressed S. aureus cells, yflE restored growth and LTA synthesis. By contrast, yfnI restored the staphylococcal growth only partially and LTA synthesis was observed but the product showed a retarded electrophoretic mobility. The other two more distant homologues, yqgS and yvgJ, did not restore growth or LTA synthesis.
When the dgkB gene was placed under the Pspac promoter, cell growth became dependent on IPTG, indicating that the dgkB repression leads to lethality (Fig. 2), as had been shown by Jerga et al. (2007). We introduced disruptions of either or both of yflE and yfnI into the Pspac-dgkB strain and found that the lethal effect was suppressed: the cells of all three transformed strains were able to grow well in the absence of IPTG (Fig. 2a). Although yfnI cannot fully substitute for ltaS in S. aureus (Gründling and Schneewind, 2007), the suppression by the yfnI single disruption (Fig. 2a) suggests that YfnI produces DG and thus synthesizes LTA in B. subtilis cells as recently shown by Wörmann et al. (2011).
 View Details | Fig. 2 Suppression of the lethality due to dgkB repression by yflE and yfnI disruptions (a) and a complementation test of the suppressive effect by ectopically expressed homologues (b). Overnight cultures in buffered LB medium containing 1 mM IPTG and appropriate antibiotics were washed, diluted to OD600=0.05 and then tenfold serially down to 10–4 with saline. One microliter each was spotted on LB agar plates containing no antibiotic with or without 1 mM IPTG (a) ; LB agar contained 1% xylose in (b). The plates were incubated at 37°C for ca. 9 h and then at room temperature overnight. Strains used in (a) were 168, YERQp, MBS13, MBS14, and MBS15. Strains used in (b) upper panel were 168, YERQp, MBS13, MBS21, and MBS22 and those in (b) lower panel were 168, YERQp, MBS14, MBS23, and MBS24. |
The growth of cells with yflE-disruption was slightly poorer (or colonies were smaller) than that of the parental Pspac-dgkB cells in the presence of the inducer. This was probably because the defect in yflE is slightly deleterious to cell growth. Poor growth of a yflE mutant has previously been reported by Schirner et al. (2009). When the Pxyl promoter-controlled yflE integrated in the amyE locus was expressed by an addition of 1% xylose, the growth of the yflE-disrupted strain became IPTG-dependent again (Fig. 2b). The yflE disruption was complemented by ectopic expression of yflE. The expression of yfnI from Pxyl promoter, however, did not cause the growth of the yflE-disrupted strain to become IPTG-dependent, i.e. cross-complementation was not observed (Fig. 2b). In the yfnI-disrupted strain, the ectopic expression of yflE from Pxyl promoter did not, while ectopic yfnI expression did return the growth to IPTG-dependent, indicating that there was no cross-complementation in the opposite direction either. The results suggest that the two gene products, YflE and YfnI, are functionally different, although the two enzymes have the same predicted membrane topology and domain structure (Wörmann et al., 2011).
Repression of dgkB in the Pspac-dgkB strain led to a markedly increased cellular level of DG, as revealed by continuous labeling with radioactive acetic acid followed by TLC analysis of the extracted lipids: three hours after IPTG removal the DG level had increased to 43 ± 3% of total lipids from a level of 26 ± 5% in the cells incubated with IPTG and very much higher than the 13 ± 1% observed in wild type cells (Fig. 3a). The higher DG level of Pspac-dgkB cells in the presence of IPTG indicates that Pspac is a weaker promoter, even when induced with 1 mM IPTG, than the native promoter for the dgkB gene. DG accumulation in dgkB-repressed cells was also shown in a pulse-chase experiment by Jerga et al. (2007). Disruption of yfnI in the dgkB-repressed cells reduced the DG content to 15 ± 2%, which is close to the wild type level, but unexpectedly yflE-disruption did not (42 ± 1%) in cells whose growth defect was noticeably suppressed to the level of the wild type (Fig. 3b). The fact that no suppression of DG accumulation was observed in yflE-disrupted cells indicates that YflE may have little effect, as opposed to YfnI, on the accumulation and further that the growth defect after dgkB repression is not directly linked to the observed DG accumulation. Wörmann et al. (2011) have reported that strains expressing YfnI in the absence of YflE in both B. subtilis and S. aureus cells produced LTA of retarded mobility (this was first observed in Fig. 4G of Gründling and Schneewind, 2007), presumably caused by an increase (× 1.6) in glycerolphosphate chain length compared with LTA produced by YflE. They suggested that YfnI becomes more efficient (or activated) in LTA synthesis when YflE is absent. The DG accumulated in yflE-disrupted cells (Fig. 3a), may thus possibly result from the efficient synthesis of LTA, yielding a large amount of DG, by the more efficient YfnI that has been freed from an as yet unknown control by YflE.
 View Details | Fig. 3 Effect on DG accumulation of disruption yflE and yfnI in the dgkB-repressed cells. Cells of the disrupted strains and their parental strain YERQp (Pspac-dgkB) were continuously labeled with [1-14C] acetic acid and harvested 3 h after removal of 1 mM IPTG (–) or without its removal (+). Cells of wild-type strain 168 were labeled in the absence of the inducer but subjected to the same washing and diluting procedures. Lipids were extracted and analyzed for DG content by TLC as described in MATERIALS AND METHODS. (a). The position of 1, 2-diacylglycerol is indicated by 1, 2-DG. The position of the 1, 3-isomer of DG, which is likely formed by spontaneous isomerization, is indicated by 1, 3-DG. The 1, 3-isomer was also detected when authentic DG (1, 2-dioleoyl-sn-glycerol; Avanti Polar Lipids) was subjected to TLC and stained with primuline. The position of fatty acids is indicated by FA. Other polar lipids (phospholipids and glucolipids) remain at the chromatographic origin indicated by O. The percentage of DG (1, 2-DG plus 1, 3-isomer) to total lipids is shown above each lane. Mean values of experiments with three different cultures are given with standard deviations. Growth of the cells of the disrupted strains and their parental strain used for the lipid extraction are shown (b). Strains 168 (*), YERQp (□, ■), MBS13 (◇, ◆),MBS14 (△, ▲), and MBS15 (○, ●) in the absence (open symbols) or presence (closed symbols) of 1 mM IPTG. |
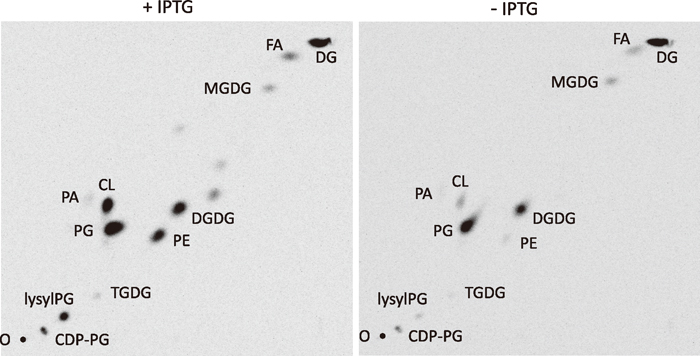 View Details | Fig. 4 Effect of dgkB repression on phospholipids. The lipid extracts of the Pspac-dgkB cells prepared as described in the legend to Fig. 3 were analyzed by two-dimensional TLC, first (x dimension) with chloroform/methanol/water (65:25:4, v/v/v) and then (y dimension) with chloroform/methanol/acetic acid (65:25:10, v/v/v), as described in MATERIALS AND METHODS. O, chromatographic origin. See legends to Fig. 1 for abbreviations. |
The results shown in Fig. 3 also indicate that each of YqgS and YvgJ makes only a small contribution to DG accumulation, although they may be partly responsible, in addition to PA phosphatase, for the DG accumulation in cells doubly disrupted for yflE and yfnI (18 ± 3%). The expression levels of the yqgS and yvgJ genes are low during vegetative growth, and these genes seem to play important roles during sporulation: a yflE yqgS-doubly disrupted strain did not sporulate, and a yflE yvgJ-doubly disrupted strain showed a reduced sporulation efficiency, while a yflE-disrupted strain sporulated normally (Schirner et al., 2009). Although YqgS has LTA-synthesizing activity (Wörmann et al., 2011), its contribution to DG production is probably very small during vegetative growth. YvgJ is likely to function as an LTA primase transferring a single G1P to the membrane anchor (Wörmann et al., 2011), which would produce a much smaller amount of DG than the LTA polymerization performed by the other enzymes.
Since DG kinase reintroduces DG into the phospholipid synthetic pathway (Fig. 1), a lower supply of PA due to dgkB repression may result in reduced synthesis of phospholipids. Reduced PG content might then lead to a growth defect as it does in pgsA-repressed cells (Hashimoto et al., 2009), although the direct cause for the defect is still unclear. Two-dimensional TLC analyses of the labeled lipid extracts of the Pspac-dgkB cells (YERQp strain) at 3 h after IPTG removal, however, showed that PG content was not reduced (31.5% to 30.9% of total lipids), while the contents of phosphatidylethanolamine (PE), cardiolipin (CL) and lysylphosphatidylglycerol (lysylPG) were greatly decreased (5.9% to 0.4%, 5.8% to 0.7%, and 6.0% to 0.6%, respectively) (Fig. 4, Table 3). Note that both CDP-DG and PA, the essential intermediates for the synthesis of phospholipids and others, were not decreased (1.8% to 2.2% and 0.4% to 0.4%, respectively). Disruption of yflE largely mitigated the decrease of PE, CL, and lysylPG (13.7% to 6.7%, 2.8% to 2.1%, and 4.9% to 3.4%, respectively). Disruption of yfnI, both in the cases with and without yflE disruption, fully mitigated the reduction of these phospholipids.
 View Details | Table 3 Effect on lipid composition of disruption of yflE and yfnI in Pspac-dgkB cells |
Mutants singly or multiply defective in the synthesis of phospholipids other than PG grow almost normally (Matsumoto et al., 1998; Kawai et al., 2004, 2006; Salzberg and Helmann, 2008). Thus, it is unlikely that the growth defect of the dgkB-repressed cells is due to interrupted reintroduction of DG into the phospholipid synthetic pathway. The result of Table 3 suggests that the imbalance of phospholipid composition induced after the interrupted reintroduction into the phospholipid synthetic pathway and simultaneous consumption of PG through LTA synthesis, which causes the large increase in DG content, but not the increase in DG content alone, might be the cause for the growth stasis of dgkB repressed cells.
The large reduction of PE content in contrast to the small PG reduction may suggest a possible lower affinity with the substrate CDP-DG of phosphatidylserine synthase, which catalyzes the committed step for PE synthesis, compared to that of phosphatidylglycerophosphate synthase, which catalyzes the committed step for PG synthesis. However, comparably large reductions of CL and lysylPG content, both synthesized downstream from PG, suggest that it is not simply ascribable to a possible difference in affinity. It seems that B. subtilis cells have a mechanism that minimizes the reduction of PG content at the expense of other phospholipids, PE, CL and lysylPG, when PA is in short supply.
Glucolipid synthase encoded by the ugtP gene processively transfers glucose from UDP-glucose to DG (Jorasch et al., 1998a), thus draining DG into the glucolipid synthetic pathway (Fig. 1). Disruption of ugtP is nonlethal in a wild-type background (Price et al., 1997; Jorasch et al., 1998b), but how deleterious is it in a dgkB-repressed background? We introduced a ugtP::kan disrupted allele into Pspac-dgkB strains carrying disruptions of either or both of yflE and yfnI. The Pspac-dgkB yflE strain became partially IPTG-dependent for its capacity to form colonies (that is, the suppression by yflE disruption was weakened) when the disrupted allele of ugtP was introduced, whereas the growth of the Pspac-dgkB yfnI ugtP strain was almost the same in the presence and absence of the inducer (Fig. 5). Inactivation by ugtP disruption of the DG-draining pathway presumably increases DG accumulation to a deleterious level upon prolonged incubation on plate in Pspac-dgkB yflE cells, which retain efficient LTA synthetic activity via YfnI (Fig. 3a).
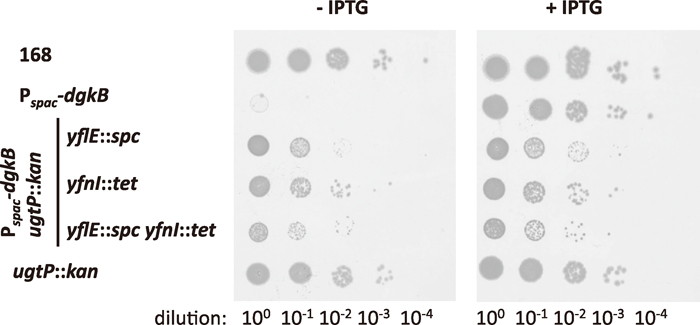 View Details | Fig. 5 Effect of deletion of ugtP on the suppression of lethality due to dgkB repression by yfnE and yflI disruption. For the experimental procedure see the legend to Fig. 2. Strains used were 168, YERQp, MBS16, MBS17, MBS18, and YTB019. |
Although the dgkB-repressed cells were viable if either or both of yflE and yfnI were disrupted, basal level expression of Pspac-dgkB in the absence of IPTG might possibly contribute to the viability. To test for this possibility, we constructed a disrupted dgkB allele and introduced it into yflE- and yfnI-disrupted strains.
First, we integrated a pMUTINCC plasmid having an internal fragment of the dgkB gene into the chromosome of a strain that harbors a gene encoding N-terminally GFP-fused DgkB under the control of Pxyl at the amyE locus in the presence of xylose, and screened the integrants by PCR for those where integration occurred at the native dgkB locus, not in the ectopic gfp-fused locus at amyE. The resultant dgkB::pMUTINCC (erm) mutant harboring amyE::(spc Pxyl-gfp-′dgkB), designated as strain TMB003, was dependent on xylose for growth.
The disrupted allele dgkB::pMUTINCC (erm) of the chromosomal DNA of TMB003 was introduced into the recipient strains disrupted for either or both of yflE and yfnI at transformation efficiencies only slightly lower than those of the introduction of the Pspac-dgkB allele in the presence of IPTG; the relative transformation efficiencies of the strains MBS10 (yflE::spc), MBS11 (yfnI::tet), and MBS12 (yflE::spc yfnI::tet) were 0.44 ± 0.2, 0.45 ± 0.11, and 0.35 ± 0.12, respectively. In the control experiment in which wild-type strain 168 was transformed with the TMB003 DNA, a few minute colonies appeared on the erythromycin-containing selection plates, but they did not continue to grow when plated on LB agar containing erythromycin. To confirm that the introduced dgkB::pMUTINCC (erm) was not coupled with the Pxyl-gfp′-′dgkB allele, we picked up eight erythromycin-resistant transformants for each recipient strain and checked whether the amyE::(spc Pxyl-gfp′-′dgkB) allele was simultaneously introduced. No such transformant was obtained, indicating that the Pxyl-gfp′-′dgkB allele was not simultaneously introduced. We conclude, therefore, that the disrupted dgkB::pMUTINCC (erm) allele was introduced into the strains with disruption of either or both of yflE and yfnI, demonstrating that these strains with abolished dgkB expression are viable.
Since PgsA, a key enzyme for the synthesis of PG, is localized to the division septum in B. subtilis cells (Nishibori et al., 2005), LTA synthases that utilize PG as a substrate were expected to show similar subcellular localization. We expressed N-terminally GFP-fused YflE and YfnI proteins at low levels (by inducing the Pxyl promoter with 0.05% xylose) from the fusion genes integrated in the amyE locus and observed that they were localized to the septum under a fluorescence microscope, as expected (Fig. 6, a and b). Septal enrichment of YflE at a higher expression level (with 0.5% xylose) was reported by Schirner et al. (2009).
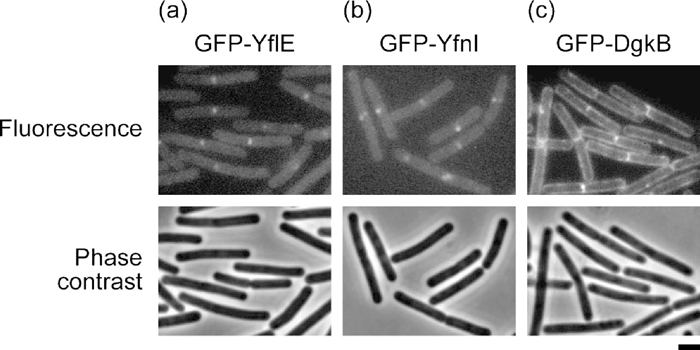 View Details | Fig. 6 Subcellular localization of GFP-YflE (a), GFP-YfnI (b), and GFP-DgkB (c) fusion proteins. The fusion genes under Pxyl control were integrated into the amyE locus of strain 168 and induced with 0.05% xylose in LB medium. Cells were harvested at ca. 100 Klett units, and fluorescence and phase-contrast micrographs were taken with exposure times of 4 s and 0.04 s, respectively. Scale bar, 1 μm. |
Since YflE and YfnI produce a large amount of DG, we expected that DG kinase would show a subcellular localization similar to these LTA synthases. However, an N-terminally GFP-fused DgkB protein expressed from the fusion gene integrated in the amyE locus was found not only in the septal region but also along the cell periphery (Fig. 6c). Although DgkB is a soluble protein (Jerga et al., 2007), it should reside on the membrane surface to phosphorylate DG in the membrane. S. aureus DgkB was demonstrated to have an affinity for anionic phospholipids and to bind to phospholipid vesicles in vitro (Jerga et al., 2009), and perhaps B. subtilis DgkB moves around for similar reasons.
In E. coli, the major source of DG is the transfer of the G1P moiety of PG to generate mature membrane-derived oligosaccharides (MDOs) (Raetz and Newman, 1979). DG kinase of E. coli is encoded by the dgkA gene. It is a membrane protein (Sanders et al., 1996) and belongs to a different protein family than the soluble DG kinase family, which includes B. subtilis DgkB (Jerga et al., 2007). Although dgkA is not an essential gene, dgkA-defective E. coli mutants do not grow well at low osmolarity (Raetz and Newman, 1978) or in the presence of arbutin (Jackson et al., 1984). At low osmolarity MDO synthesis is greatly enhanced (Kennedy, 1982). Arbutin is an analog of MDO in terms of G1P transfer from PG, and in its presence the transferase shows high activity even at high osmolarity (Bohin and Kennedy, 1984). Consequently, under these conditions, a large amount of DG accumulates. Thus it would appear that DG accumulation is deleterious to cell growth. It is, however, yet to be elucidated why DG accumulation is deleterious to bacterial growth.
We show here that disruption of yflE and yfnI, or of one of these LTA synthesis genes suppresses the lethality of dgkB repression and that the accumulation of DG after dgkB-repression was suppressed by the disruption of yfnI, indicating that the DG accumulation is involved in the lethality after dgkB repression. However, yflE disruption did not suppress the DG accumulation while it did suppress the growth defect, indicating that DG accumulation is not deleterious to the yflE-disrupted cells. The DG accumulation in yflE-disrupted cells, presumably resulted from the efficient synthesis of LTA by YfnI freed from a possible control by YflE (Wörmann et al., 2011), but we cannot explain the reason for the suppressive effect of the yflE disruption. Further disruption of ugtP, whose product consumes DG as a substrate (see Fig. 1), partially weakened the suppression by yflE disruption. We conclude that DgkB is required to keep the cellular level of DG below a lethal level and further that disruption of either or both of yflE and yfnI makes dgkB dispensable. Indeed, a disrupted dgkB allele was successfully introduced into these disruptants.
Only minor DG accumulation was detected in cells doubly disrupted for yflE and yfnI, indicating that YqgS and YvgJ make an even smaller contribution to DG production than YflE and YfnI during vegetative growth. LTA synthesis by YflE and YfnI is most likely a major source of DG production in the vegetative cells of B. subtilis. The role of YflE is believed to be that of a “housekeeping” enzyme, while YfnI may be a “stress-responding” enzyme as the latter is possibly sigma M-dependent (Wörmann et al., 2011; Sutcliffe, 2011). YfnI becomes more efficient in LTA synthesis and produces longer polymers (× 1.6 in chain length) than YflE when YflE is absent, suggesting that the activity of YfnI is under some sort of control by YfnE. The exact roles the enzymes play in vivo and the precise relationships between them remain to be elucidated.
DG accumulation in the dgkB-repressed cells has a deleterious effect on B. subtilis growth as it does in the case of E. coli. However, DG accumulation in yflE-disrupted cells does not. This obviously contradictory result indicates that we are far from a full understanding of the essential role that DgkB plays in the DG cycle and that of LTA on the cell surface of B. subtilis. The longer LTA polymer on the surface of yflE-disrupted cells may have a function that mitigates the deleterious effect of DG accumulation. The analysis with two-dimensional TLC has revealed an imbalance of phospholipid composition in dgkB-repressed cells (Table 3). The imbalance induced after the interrupted reintroduction of DG into the phospholipid synthetic pathway and simultaneous consumption of PG through LTA synthesis, which causes DG accumulation, might be the cause for the growth stasis of dgkB repressed cells.
We are grateful to Kei Asai and Yu Tanimura for bacterial strains, and Atsushi Yamashita for his invaluable advice on lipid analysis. We are also grateful to Yoshito Sadaie, Yosuke Koga, and Hiroaki Takahashi for their encouragement and helpful discussions. This study was supported in part by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science.
|